
单核细胞增生李斯特菌LadR蛋白对外排泵MdrL的调控机制研究
2018-12-25 11:12:30徐雅梦姜晓冰于涛
生物技术通报 2018年12期
徐雅梦 姜晓冰 于涛
(1. 河南师范大学生命科学学院,新乡 453007;2. 新乡学院生命科学技术学院,新乡 453003)
单核细胞增生李斯特菌(Listeria monocytogenes,Lm)是一种重要的食源性致病菌,该菌在自然界分布广泛,易对熟肉制品、乳制品、海产品和蔬菜等食品造成污染。摄入被Lm污染的食品可引起李斯特菌病(Listeriosis)的爆发和流行[1]。老人、孕妇、新生儿以及免疫力低下者是李斯特菌病的易感人群,临床症状主要表现为败血症、脑膜炎、孕妇流产等[2]。李斯特菌病在全球范围内的发病率较低,但是其致死率却高达25.9%[3],严重威胁人类的健康。为了防止食品被致病菌污染,食品企业通常使用消毒剂来抑制或杀灭食品加工环境中的微生物。苯扎氯铵(Benzalkonium chloride,BC)属于季铵盐类消毒剂,被广泛应用于食品工业领域。BC的长期使用可能会促使耐药菌株的出现,影响其消毒效果。
许多文献报道,从食品加工环境和市售食品中分离的Lm菌株对BC敏感性下降[4-6]。目前研究者普遍认为外排泵是介导Lm对BC耐受的主要机制。外排泵属于膜转运蛋白,在细菌中广泛存在。除了参与细胞的正常物质运输和代谢,外排泵还能够去除细菌细胞和胞膜中的有害物质,帮助细菌抵御外界不良环境的影响[7]。研究报道,一些外排泵可将抗生素、消毒剂、重金属等作为底物排出菌体外使细菌产生耐药性[8]。我们先前的研究发现,主要易化子超家族(Major facilitator superfamily,MFS)外排泵MdrL在Lm对BC耐受中起作用。根据Lm全基因组序列可知,ladR基因反向位于mdrL外排泵基因的上游,编码的LadR蛋白由176个氨基酸分子组成,属于PadR转录负调控蛋白家族。Huillet等[9]推测,mdrL基因的启动子区域可能存在调控蛋白LadR的结合位点,但这一推测并没有被进一步证实。本研究以Lm标准菌株EGD-e为研究对象,利用同源重组技术[10]构建ladR基因缺失菌株ΔladR,通过实时荧光定量PCR(Quantitative reverse transcriptase-polymerase chain reaction,qRT-PCR)、凝胶阻滞试验(Electrophoretic mobility shift assay,EMSA)等调查LadR蛋白对外排泵MdrL的调控作用及调控方式。
1 材料与方法
1.1 材料
1.1.1 菌株及质粒 Lm标准菌株EGD-e由华中师范大学罗勤教授惠赠;穿梭质粒pMAD和表达载体pET32a由本实验室保存;大肠杆菌DH5α和BL21(DE3)感受态细胞购自北京博迈德基因技术有限公司。
1.1.2 培养基及主要试剂 脑心浸液培养基(Brain Heart Infusion,BHI),购自北京陆桥生物技术有限公司;苯扎氯铵,购自上海阿拉丁生化科技股份有限公司;细菌基因组DNA提取试剂盒和质粒小提试剂盒,购自北京天根生化科技有限公司;胶回收试剂盒、PCR产物纯化试剂盒、T4连接酶、限制性内切酶,购自美国Thermo Fisher;TaqDNA聚合酶和dNTP,购自大连宝生物工程有限公司;His标签蛋白纯化试剂盒、BCA蛋白浓度测定试剂盒、EMSA/Gel-Shift结合缓冲液(5×),购自碧云天生物技术研究所;引物由上海捷瑞生物工程有限公司合成。
1.1.3 仪器与设备 DH360型电热恒温培养箱,北京科伟永兴仪器有限公司;THZ-82气浴恒温振荡器,金坛天竟实验仪器厂;TGL-16B型离心机,上海安亭科学仪器厂;ETC811型基因扩增仪,北京东胜创新生物科技有限公司;DYY-8C型电泳仪,北京市六一仪器厂;LightCycler@96实时荧光定量PCR仪,瑞士Roche;UNIVERSAL HOOD II 凝胶成像分析系统、Gene PulserXcellTM电转仪,美国 Bio-Rad。
1.2 方法
1.2.1ladR基因缺失突变株的构建与分子鉴定 以EGD-e基因组DNA为模板,分别用1408-1/1408-2和1408-3/1408-4两对引物扩增ladR基因的上下游同源臂,引物序列见表1。利用重叠延伸PCR(Splicing by Overlap Extension,SOE-PCR) 技 术[11]融 合 上下游同源臂,融合片段经切胶回收后与穿梭载体pMAD分别用限制性内切酶BamH I和MluI进行同步双酶切。酶切产物经纯化后进行连接反应,将构建好的重组质粒pMAD-ΔladR转化至大肠杆菌DH5α中,将阳性转化株送至北京博迈德基因技术有限公司进行测序。
采用青霉素G法制备EGD-e感受态细胞[12]。将测序正确的重组质粒pMAD-ΔladR与EGD-e感受态细胞混匀,电击后立刻加入500 μL BHI培养基于30℃培养3 h。将菌液均匀涂布于含红霉素(5 μg/mL)、和X-gal的BHI平板上,30℃培养48 h。挑取蓝色单菌落于含红霉素(5 μg/mL)的BHI液体培养基,39℃培养24 h。稀释菌液选择合适梯度的稀释液涂布于含红霉素(5 μg/mL)和X-gal的BHI平板上,39℃培养48 h。挑取蓝色单菌落于BHI液体培养基,30℃培养24 h。将菌液转接至新鲜的BHI,39℃培养。稀释菌液选择合适梯度的稀释液涂布于含X-gal的BHI平板上,39℃培养48 h。挑取白色单菌落用引物1408-1和1408-4进行PCR扩增检测缺失条带,最终获得ΔladR突变株。

表1 PCR扩增所用引物
1.2.2mdrL基因转录水平检测 将野生株EGD-e和突变株ΔladR分别接种至BHI中(各6管),37℃振荡培养至OD600nm≈0.6。将菌株平均分为两组,第一组为不加任何药物(作为对照组),第二组为加入BC(2 μg/mL),37℃继续培养30 min。立即加入10%的反应终止液(苯酚∶无水乙醇=1∶9)[13],冰上放置10 min。
按照细菌RNA提取试剂盒的使用说明提取细菌总RNA,测定RNA的浓度和纯度后用于后续试验。利用cDNA第一链合成试剂盒将RNA反转录为cDNA,-20℃保存备用。根据GenBank的基因序列设计靶基因mdrL和参照基因16S rRNA的引物(表2)。以cDNA作为模板,使用RealMasterMix(SYBR Green)试剂盒进行 qRT-PCR。采用 ΔΔCT法[14]进行目的基因表达量的分析。
1.2.3ladR基因的克隆 以EGD-e的基因组DNA作为扩增模板,用引物ladR-q1和ladR-q2(表1)扩增ladR全基因序列。ladR扩增产物与表达载体pET32a分别用限制性内切酶BamH I和XhoI进行同步双酶切,酶切后的产物经纯化后进行连接反应,将构建好的重组表达载体pET32a-ladR转化至大肠杆菌BL21(DE3),挑选阳性转化株送至北京博迈德公司测序。
1.2.4 LadR蛋白的表达与纯化 将LadR蛋白表达菌株接种于5 mL含Amp(100 μg/mL)的LB液体培养基,37℃过夜培养。过夜培养物按1∶100分别接种至50 mL含Amp(100 μg/mL)的LB液体培养基,37℃振荡培养至OD600nm≈0.6。将菌液分两组,一组不做任何处理,另一组加入终浓度为0.5 mmol/L[15]的 IPTG,37℃ 继 续 培 养 8 h。 离 心(4 000×g,5 min)收集细胞。按照His标签蛋白纯化试剂盒的步骤提取纯化重组蛋白His6-LadR。取10 μL纯化蛋白进行SDS聚丙烯酰氨凝胶电泳(SDS-Polyacrylamide gel electrophoresis,SDS-PAGE)[16]。利用 BCA 蛋白浓度测定试剂盒测定重组蛋白His6-LadR的浓度。
1.2.5 凝胶阻滞试验 利用引物pmdrL-F/pmdrL-R扩增mdrL的启动子区域,同时随机挑选一段编码区DNA(recA)作为阴性对照。将纯化后的DNA(200 ng)与纯化后的重组蛋白His6-LadR在EMSA/Gel-Shift结合缓冲液中混合,室温放置20 min。利用6%非变性PAGE电泳进行结合活性检测[17]。
2 结果
2.1 ladR基因缺失菌株的构建与分子鉴定
用引物1408-1/1408-2扩增得到ladR基因上游同源臂480 bp,用引物1408-3/1408-4扩增ladR下游同源臂168 bp。通过SOE-PCR得到648 bp的上、下游同源臂融合片段ΔladR,条带均与预期结果一致(图1-A)。融合片段ΔladR与穿梭质粒pMAD连接获得重组质粒pMAD-ΔladR。双酶切验证结果如图1-B。
测序正确的重组质粒pMAD-ΔladR电转至EGD-e感受态。筛选得到的阳性菌株用检测引物1408-1/1408-4只能扩增出648 bp的片段(图1-C)。测序结果表明已成功缺失ladR基因,获得ΔladR突变株。
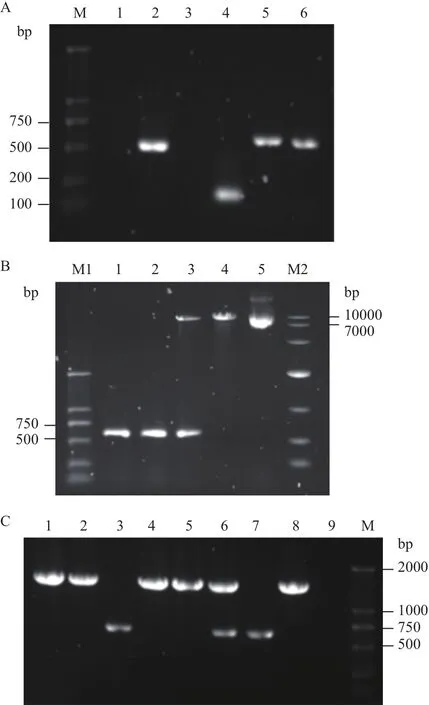
图1 ladR基因缺失株的构建
2.2 mdrL基因转录水平分析
由图2可明显看出,与野生株相比,突变株ΔladR的mdrL基因的转录水平提高约51倍。BC胁迫下,野生株EGD-e的mdrL基因转录水平提高约1.6倍,突变株ΔladR的mdrL基因的转录水平约1.4倍。该结果表明LadR对mdrL基因有负调控作用。
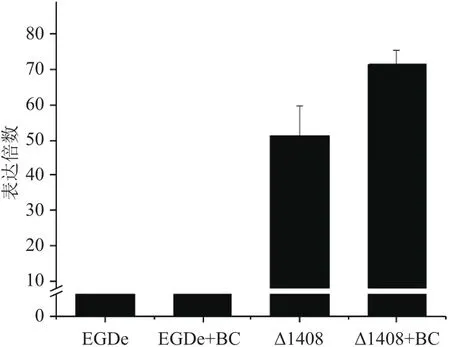
图2 qRT-PCR检测mdrL基因转录水平
2.3 ladR基因的克隆
以EGD-e基因组DNA为模板扩增得到长度为525 bp的ladR全基因片段(图3);构建得到的重组质粒pET32a-ladR经双酶切验证及测序,验证结果一致表明已成功获得表达菌BL21(DE3)-ladR。
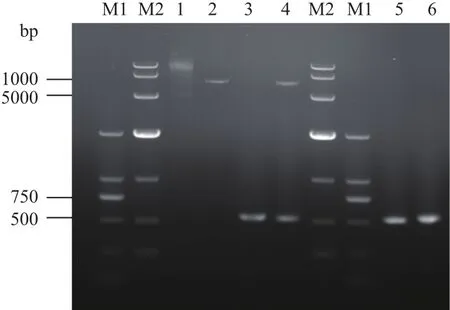
图3 表达菌BL21(DE3)-ladR的构建
2.4 LadR蛋白的表达及纯化
IPTG诱导后,表达宿主菌在35 kD附近出现一条明显的蛋白条带,与预期的蛋白分子量大小(36.5 kD)十分相近(图4),表明目的蛋白在宿主菌已得到成功表达。利用His标签蛋白纯化试剂盒处理后可以得到高纯度的His6-LadR蛋白;经BCA蛋白浓度测定试剂盒测定可知其浓度为1 mg/mL。
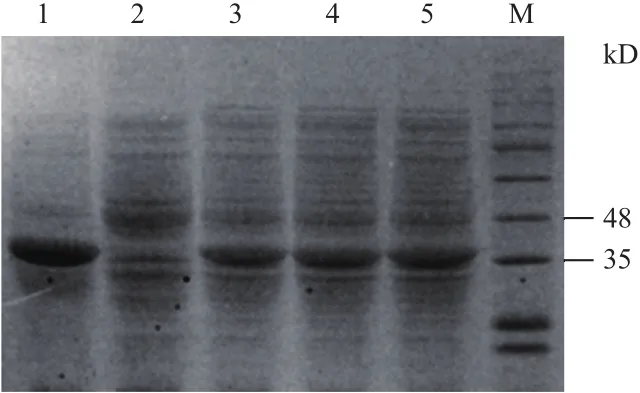
M:蛋白maker;1:纯化蛋白;2:阴性对照;3~5:IPTG诱导
2.5 凝胶阻滞试验
EMSA结果(图5-A)显示,LadR蛋白可与mdrL基因启动子区域结合,并且该结合作用在一定范围内随LadR蛋白浓度升高而增强;而在所有蛋白浓度下,阴性对照recA基因均未出现阻滞条带(图5-B)。结果表明,LadR蛋白可与mdrL启动子序列发生特异性结合作用。
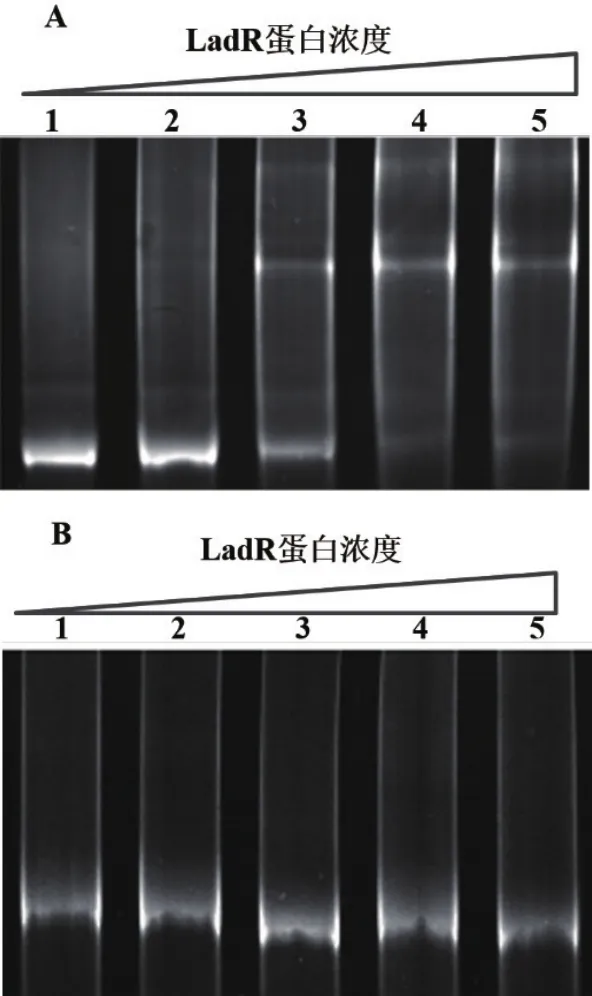
图5 重组蛋白His6-LadR与启动子区域结合活性
3 讨论
Tamburro等[18]研究报道,BC胁迫下mdrL基因出现明显过表达现象,推测MdrL外排泵在Lm对BC耐受中起作用。Romanov等[19-20]通过检测不同Lm菌株中mdrL基因的表达量发现,BC胁迫下mdrL基因的转录水平升高4-20倍,该结果表明在BC刺激下不同菌株的mdrL基因表达量呈现差异性。我们先前的研究结果显示,与野生株EGD-e相比,mdrL基因缺失突变株ΔmdrL在亚致死浓度BC胁迫下的生长迟滞期延长、平均最大生长率和平均最大光密度值均降低;在致死浓度BC作用下的存活率降低2个log值,表明MdrL外排泵在Lm对BC耐受中起作用。本研究结果显示,BC作用下,mdrL基因在野生株EGD-e中的转录水平提高约1.6倍,表明BC能够诱导mdrL基因的表达。不过,与前人的研究结果相比,本研究中mdrL基因转录水平升高的倍数较低。其原因可能有三点:一是菌株自身的差异性;二是BC刺激的时期不同;三是BC作用的浓度不同。
Huillet等[9]利用 Northern blot技术检测mdrL基因的表达水平,结果发现LadR蛋白对mdrL基因具有负调控作用,并推测在正常生长条件下(BHI培养基),LadR与mdrL基因的启动子区域结合,从而抑制mdrL外排泵基因的转录。本论文在前人研究的基础上,深入调查LadR蛋白对MdrL外排泵的调控机制。qRT-PCR结果显示,无BC刺激时,mdrL基因在ladR基因缺失突变株中的转录水平比野生株EGD-e高51倍,表明LadR蛋白可能负调控mdrL基因的转录;BC作用下,mdrL基因在突变株中的转录水平比EGD-e高1.4倍,我们推测在BC胁迫下,除了LadR蛋白可能还存在其他的调控蛋白参与mdrL的调控。EMSA试验[21]结果显示,LadR蛋白能够与mdrL基因启动子区域结合,并且其结合活性随蛋白浓度的增加而增强,当蛋白浓度大于0.4 mg/mL时,结合活性没有明显变化。LadR蛋白与recA基因的编码区没有发生结合作用,表明LadR与mdrL基因启动子区域的结合具有特异性。
4 结论
本研究以Lm标准菌株EGD-e为研究对象,深入调查LadR对MdrL外排泵的调控作用及其调控方式。结果表明,BC能够诱导mdrL基因的表达。LadR蛋白负调控mdrL基因的表达;在正常生长条件下,LadR与mdrL基因的启动子区域结合,抑制外排泵基因mdrL的表达。
猜你喜欢
工程与建设(2019年3期)2019-10-10 01:40:36
食品科学(2018年10期)2018-05-23 01:27:28
基础医学与临床(2018年2期)2018-02-12 13:12:30
科技创新导报(2016年31期)2017-03-30 09:15:27
法医学杂志(2015年4期)2016-01-06 12:36:36
法医学杂志(2015年4期)2016-01-06 12:36:36
西南医科大学学报(2015年1期)2015-08-22 13:01:46
中国当代医药(2015年9期)2015-03-01 02:01:59
西南军医(2015年6期)2015-01-23 01:25:50
当代畜禽养殖业(2014年6期)2014-02-27 07:59:03
