
分子动力学模拟在环氧基复合材料中的应用
2018-12-21 02:57徐喻琼
粘接 2018年12期
徐喻琼
(三峡大学机械与动力学院,湖北 宜昌 443002)
环氧树脂是一类分子结构中含有至少2个以上环氧基团的聚合物,可与含有活泼氢的胺和酐进行反应、固化交联生成网状结构。环氧基复合材料因具有高模量、低蠕变和优良力学性能而广泛用于涂层、胶粘剂和电子航空领域[1,2]。
有研究表明[3]高模量的环氧树脂基体是决定环氧基复合材料具有优良力学性能和界面性能的重要因素。由于生成复合材料的树脂和固化剂有多种化学结构,而且交联网络非常复杂,因此,对环氧基复合材料的实验研究非常耗时且效率极低。
长期以来,研究人员一直希望能够根据环氧树脂的模量、固化剂的特性预测材料的性能、了解材料的微观结构,进而对环氧基复合材料的固化剂、纳米填料等进行选材和设计。而分子模拟方法是以原子—分子模型为基础的,在研究分子结构和相互作用方面具有较大的优势[4]。在研究环氧树脂基复合材料方面,分子模拟方法不仅可以模拟分子的静态结构,还可以模拟分子的动态行为,为研制具有更优越综合性能的环氧基复合材料提供了一种新的研究模式,大大加速了产品的设计和研发过程[5]。
1 交联环氧树脂的模型
由于脂环有一定的空间位阻,与环氧树脂在室温下只能进行加成开环反应,完全固化要依靠加热。环氧树脂与伯胺的反应机理得到了较多地研究,其包含3个主要的反应[6]:环氧基与伯胺开环加成反应生成仲胺;仲胺与环氧基反应生成叔胺;在高温或催化剂存在下,生成的仲胺和叔胺在加成物上的羟基可以与环氧基继续反应。图1为环氧树脂与伯胺的反应机理。
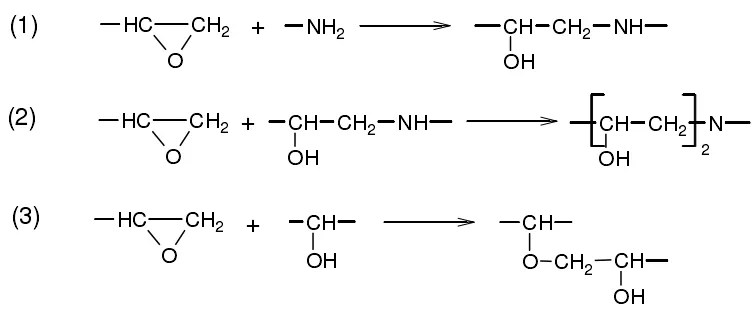
图1 环氧树脂与伯胺的反应机理Fig.1 Reaction mechanism of epoxy resin with primary amine
分子动力学是研究不同物质分子行为的一种数值模拟技术,其模拟流程如图2所示。模拟过程有如下步骤:①建立原子几何模型;②定义原子间的相互作用;③设定系统控制方程;④初始化及能量最小化;⑤设定模拟条件;⑥整合方案;⑦计算性能[7]。
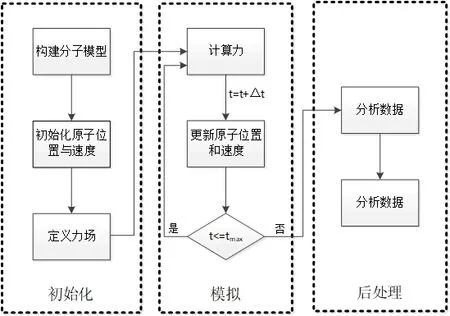
图2 分子动力学模拟的流程图Fig.2 Flow chart of molecular dynamics simulation
2 分子动力学模拟的有效性
Yarovsky等[8]提出了一个复杂的模型化交联系统的方法,研究了2个水性环氧体系的交联密度、收缩率、渗透性和与表面黏合性等在涂料应用中几个主要性质。其交联体系是低分子质量的,且环氧树脂和胺类固化剂的反应机理并不代表广泛的环氧基用氨基开环的机理。
Wu等[9]基于交联反应的原理,提出了纯交联环氧树脂(EP/pure)的模型,将50个双酚A二缩水甘油醚(DGEBA)和25个3'3-二氨基二苯砜(33DDS)放置入周期性盒子中,对参与反应的原子进行标记,设定反应距离,然后进行动力学弛豫,判断在反应距离内是否出现成对的反应原子,若是,则进行交联反应,连接成对原子,弛豫后增大反应距离,若否,直接增大反应距离,再次循环判断,一直到达到相对最大反应距离或目标交联度才停止。Zhu等[10]模拟了单壁碳纳米管增强环氧树脂(EPON862)复合材料的应力—应变关系和纵向弹性模量。
Li等[11]就DGEBA/33DDS环氧体系热机械性能的分子动力学计算值和实验值作了全面比较,验证了模拟结果的有效性,指出了对玻璃态环氧体系的弹性模量、热膨胀和比热等都可以定量预测,玻璃化转变温度(Tg)修正冷却速度后也可以进行精确地预测,但橡胶态时的黏弹性和热性能有待进一步地深入研究。以上这些研究结果都说明分子动力学模拟是环氧基复合材料合理选材的有效方法。
分子动力学在环氧基纳米复合材料的性能分析中也显现出显著的优点。Yu等[12]通过分子动力学的方法研究了不同尺寸的纳米氧化铝颗粒对环氧树脂基复合材料机械特性的影响,通过与实验对比和分析,小尺寸的纳米颗粒能够有效强化纳米复合材料。张晓星等[13]采用分子动力学的方法建立了二氧化硅/环氧树脂的复合模型,从微观角度分析了SiO2掺杂及其表面修饰对环氧树脂性能的影响。
Gou等[14]用分子动力学研究了EPON862的界面粘接。Choi等[15]研究了交联环氧基纳米复合材料的Tg、热弹性以及填料的分散性。Khare等[16]探究了功能化碳纳米管对环氧树脂基复合材料机械性能和热性能的影响,通过反应分子动力学仿真发现,添加氨基功能化的碳纳米管之复合材料具有更高的热导率和更好的力学性能。
分子动力学模拟提供的原子模型从底层描述了环氧基复合材料及其连接结构材料性能和平衡、非平衡的动态求解过程。模拟技术在工程结构问题中的应用还可以解释为什么在潮湿环境下存在环氧基纤维多层材料的界面脱粘等问题,但更基本的原理需要材料化学性能和结构的组合描述[7]。分子动力学虽然在分子级别上能有效地研究体系的热机械性、力学性能和耐久性,但还应该在纳米、宏观尺度和各种手段(如分子动力学、有限元模拟)等多种技术的综合应用方面予以关注。
与依靠不断开发自然资源来增加经济效益的传统模式不同,循环经济是在依靠现有自然资源的基础上,在利用资源的每一个步骤上都充分发挥资源的作用。循环经济把自然资源的开发、分解、消化作为一个循环的过程,使自然资源与生态系统有机的结合,每一个环节都能得到相应的经济效益。循环经济的特点是低污染、高利用、可持续发展[1]。
3 分子力场
力场是通过经验性的算法对体系中的势能之函数进行拟合,一般都是将原子的坐标作为势能函数的变量。分子中原子间的相互作用是通过势能函数(即力场)表示的。如果分子中原子的相对位置及其成键情况确定了,该分子系统中原子间的作用力也就确定了,这就可以对不同的模拟体系采用不同的势能函数进行表示。
分子力场在进行分子动力学模拟时,其选择性将直接关系到计算结果的准确性,环氧树脂模拟中主要有Dreiding、COMPASS、PCFF(polymer consistent force field)、ReaxFF(reactive forcefield) 和CVFF(consistent valence force field)等力场。
Wu等[9]用一般的Dreiding2.21力场和改进的Compass力场建立模型并预测了材料性能,产生了一个转变率高达93.7%的环氧-胺聚合物网络模型,根据模型的平衡结构计算了系统的密度和弹性常数,模拟结果与实验数据相吻合,该方法可用于环氧树脂材料或其他固化体系。
杜灵根[17]基于COMPASS分子力场,利用分子动力学模拟方法建立了低固化度交联耦合的双酚A型环氧树脂交联结构模型,并利用环氧树脂交联体系模型模拟计算了交联环氧树 脂 的Tg。 徐 作 瑞 等[18]采 用COMPASS和QEq电荷赋值方法,手动建立化学键,通过不断执行优化构型和搜索成键的过程,最终形成了交联度为80%的环氧树脂交联体系的分子动力学模型。在构建好的环氧树脂无定形体系中插入变化范围为0~12.5%的水分子,从而模拟了加速老化试验中不同的湿气氛围,发现模拟过程中不断有水分子脱离无定形体系并出现在体系上方的真空层中。由于水分子有较大的极性,因此容易在环氧树脂内部与体系生成氢键而较难驱离,环氧树脂的空间交联网状结构有利于阻隔水分子进入环氧树脂内部,这2者共同作用影响了环氧树脂体系的含水量。利用环氧树脂交联体系模型模拟计算了不同温度下交联环氧树脂的密度和二面角扭转能,以此预测了环氧树脂的Tg[19]。
PCFF力场适用于聚合物和有机材料,可计算内聚能、机械性能、压缩性能、热性能和弹性常数。Fan等[20]用PCFF力场模拟了交联环氧树脂的周期性无定形结构,由不同温度下的结构立体基阵进行连续堆积,计算出了Tg、线性热膨胀系数和弹性模量。
ReaxFF力场已被广泛用于碳基系统中,其 能 准 确 地 描 述 材 料 的 特 性[21,22]。Odegard等[23]证实ReaxFF已有的参数设置能成功地用于预测交联环氧树脂材料的结构和弹性响应,建立的环氧体系模型能预测材料的刚度和屈服强度。韩智云等[24]基于ReaxFF力场对设计的环氧复合材料做密度优化,得到比较符合实际的合理空间结构。
Sun等[25]用Dreiding力 场 和PCFF力 场 建立了交联环氧树脂DGEBA模型, 由Me-THPA 和DMP-30模型计算了2种力场下交联环氧树脂在室温下的径向函数分布、均方位移以及环氧树脂和石墨烯的相互作用能。结果表明,2种力场都能模拟环氧体系,但采用PCFF力场计算出的势能和密度更大,Dreiding力场更符合实验值。
此外,Yaphary等[26]选用CVFF(Consistent Valence Force field)力场模拟了NaCl溶中环氧树脂和二氧化硅界面,局部电荷由键增量方法进行估算。结果表明,宏观的耐久性行为来源于原子级别的退化,为界面耐蚀性的树脂材料合成和设计提供了参考。
4 固化剂的选择
通过选用合适的固化剂,或者在复合材料中加入纳米填料,可以得到高模量的环氧基复合材料[27~29]。但是由于固化剂化学结构的多样性,且化学结构的微小差别会带来特殊使用条件下性能的巨大差异[30],因此,选择合适的固化剂是一项艰巨的工作,通过分子动力学理解固化剂的微观结构变化对固化环氧树脂的模量影响尤为重要。
李楚新等[31]提出采用一个分子动态模拟方法预测不同结构的固化剂对环氧树脂Tg的影响。Li等[32]用分子动力学方法研究了热固性环氧基复合材料EPON862/二乙基甲苯二胺(DETDA)的热力学性能。Bandyopadhyay等[33]研究了交联密度和交联环氧树脂的热力学性能。
在选用DGEBA作为环氧树脂单体、三乙烯(TETA)为固化剂时,TETA胺基团中的氢原子与DGEBA树脂中的环氧基形成共价键,这是高 交 联 的 环 氧 结 构[34]。 Alian等[35]模 拟TETA固化DGEBA,研究了碳纳米管环氧复合材料的原子界面性能和横向各向同性的典型体积单元(representative volume element,RVE)。Jeyranpour等[36]用分子动力学比较了固化剂的效果,全面研究了DGEBA/TETA和DGEBA/DETDA环氧体系在不同交联密度、不同温度下的密度、Tg、热线胀系数,弹性模量、体积模量、剪切模量和泊松比等弹性性能均与实验值较一致。Soni等[37]用4种链长不同的聚氧化丙烯POP二元胺对DGEBA环氧单体进行交联,用模拟退火聚合法建立了交联结构,根据体积—温度关系研究了4种交联体系的密度、体积热线胀系数和Tg。由于考虑了快速冷却速率,故Tg值高于实验值,且差值随POP二元胺的链长而改变。
为了再现环氧树脂和固化剂间的化学反应,Okabe等[38]将化学反应的活化能和生成热引入固化模拟中,然后用分子动力学模拟交联固化结构,进行了固化模拟,从而推导出密度和弹性模量,结果表明,交联结构与DSC实验以及力学性能的结果相吻合。模拟结构表明,静电作用对弹性模量至关重要,羟基中的氧和氢原子形成的氢键是导致不同树脂弹性模量差异的主要因素。
Zhang等[39]通过改变固化剂二元胺的化学结构和添加单壁碳纳米管,改变了环氧基材料的模量,用原子模拟技术研究了结构—性能的关系。结果表明,改进固化剂微观结构参数会引起材料的模量变化。当预测和分析树脂交联体系及其复合材料与微观参数有关的性能时,分子动力学方法能显现出较大的价值。
5 结论
(1)分子动力学方法可将环氧基复合材料模型化,并可模拟固化的交联结构,并从微观计算预测出材料的性能,这是环氧基复合材料合理选材的有效方法。
(2)分子动力学模拟环氧基复合材料的力场有Dreiding、COMPASS、PCFF(polymer consistent force field)和ReaxFF(reactive forcefield)等。研究人员对已有力场的参数设置进行改进,从中预测了环氧基材料的力学性能和Tg等,并与实验值相吻合。
(3)分子动力学可以模拟环氧基复合材料的交联固化结构,并可进行固化模拟,根据固化剂的微观结构变化预测固化环氧树脂的密度、热力学性能及Tg。
分子动力学可以对环氧基复合材料的固化交联过程、热机械性能等进行原子尺度的微观分析,研究材料体系的性能和物理—化学机制,但材料界面处的应力—应变行为、特殊环境下的力学退化行为仍需进一步探索。如何将宏观——介观——微观多尺度的协同效应用于研究复合材料增强的内在机理是今后有待解决的科学问题。
猜你喜欢
大电机技术(2022年5期)2022-11-17
河北地质(2022年2期)2022-08-22
建材发展导向(2022年12期)2022-08-19
中国铸造装备与技术(2022年3期)2022-05-27
中国音乐学(2022年1期)2022-05-05
绿色环保建材(2021年8期)2021-09-02
世界农药(2019年4期)2019-12-30
教学与管理(理论版)(2017年7期)2017-08-11
筑路机械与施工机械化(2017年6期)2017-07-10
航天制造技术(2016年6期)2016-05-09
