
黄淮麦区部分小麦品种(系)重要产量性状全基因组关联分析
2018-12-06 02:01赵丹阳王卫东张思妮夏雪姣翟晓光马翎健
麦类作物学报 2018年11期
赵丹阳,朱 婷,王卫东,张思妮,夏雪姣,翟晓光,丁 勤,马翎健
(西北农林科技大学,陕西杨陵 712100)
基因关联分析是以连锁不平衡为基础,对自然群体中目标性状的遗传变异与基因多样性进行关联,筛选与表型变异相关分子标记的一种策略,具有低耗费、高精度、强实用性等优点,目前已广泛应用于水稻[1]、小麦[2]、玉米[3-6]、油菜[7]等作物遗传育种研究。近年来,小麦的关联分析研究主要围绕株高、穗粒数、可育小穗数、千粒重等产量性状进行。如Wu等[8]对黄淮麦区175份小麦的株高、产量相关性状进行了关联分析,从中筛选出23个显著关联的SSR标记。有研究发现,Xwmc134与小麦株高相关,在WMC363~Xgwm234之间含控制穗长的QTL位点,Xwmc710、Xbarc235和Xbarc252能以高频率从亲本遗传于后代,且与穗粒数、产量紧密相关,Xubc873a~Xwmc63区间内有与可育小穗数相关的QTL位点,Wmc48与粒重相关[9-13]。此外,Mir等[14]还发现了11个与小麦粒重关联的新型分子标记。但是,关于小麦株高、穗长、单株穗数、可育小穗数、穗粒数、千粒重等多个重要产量性状与分子标记的关联分析研究仍较少。
本研究主要以52份黄淮麦区小麦品种(系)为材料,对6个重要产量性状进行两年多点表型鉴定,利用覆盖小麦21条染色体全基因组的226个SSR标记和22个SNP标记进行关联分析,以期为小麦产量性状相关等位变异的鉴定挖掘及分子标记辅助育种提供参考。
1 材料与方法
1.1 试验材料
供试的52份小麦材料中,包括24个黄淮麦区主栽品种和28个西北农林科技大学近期培育的小麦品系。其中,黄淮麦区主栽品种中,陕西有7个,河南有8个,山东有5个,河北有2个,安徽和江苏各有1个(表1)。
1.2 表型鉴定
2015年将52份供试材料种植于陕西杨凌西北农林科技大学和河南南阳农科院试验站。2016年将供试材料分别种植于陕西杨凌西北农林科技大学试验站、江苏宿迁中江种业育种基地与河南驻马店农科院试验站。按照完全随机区组设计,单行种植,行长、行距、株距分别为1.5 m、20 cm、10 cm,每行点播15粒种子,并进行常规田间管理。2016年5月下旬于陕西杨凌、河南南阳和2017年5月下旬于陕西杨凌、河南驻马店、安徽宿迁调查供试材料表型性状。每地每个材料随机选取5个单株测量其株高、穗长,并对单株穗数、可育小穗数及穗粒数进行计数。收获时每行随机收取10个单穗,脱粒、烘干、去除碎粒后称量得其千粒重。
1.3 标记选择及基因型鉴定
通过参考文献和GrainGenes数据库(http://avena.pw.usda.gov/GG2/index. shtml)筛选出覆盖小麦全基因组的365对SSR引物,初筛后最终获得226对多态性引物(A基因组55个,B基因组73个,D基因组98个)。通过查阅文献和NCBI数据库筛选89对SNP引物对供试小麦材料进行KASP(kompetitive allele specific PCR)基因分型,筛选出多态性高的22对SNP引物(A基因组10个,B基因组5个,D基因组7个)。
2016年11月在陕西杨凌小麦育种中心实验基地取小麦嫩叶,采用改良CTAB法[15]提取DNA,DNA提取液进行质控后稀释备用。
SSR标记鉴定步骤如下:(1)SSR实验操作程序和PCR扩增参照简化的SSR分析体系[16]操作;(2)扩增产物用6%非变性聚丙烯酞胺凝胶(polyacrylamide gelelectrophoresis,PAGE)分离,以改进的银染方法[17]染色并于凝胶成像仪中观察读带;(3)分析每对SSR引物胶图,因为在150~300 kb间条带多样性丰富,从此区间读取3条多态性带,并按照软件DataFormater“0,1”带型格式[18]整理SSR信息。SNP标记鉴定均参照AS-PCR[19]扩增及产物检测的方法,用384孔板进行高通量PCR,于微孔板荧光分析仪得到所需数据。由于SNP引物较少,且其分型结果与SSR 标记格式不同,后续分析将其转化为SSR的“0,1”格式进行软件分析。
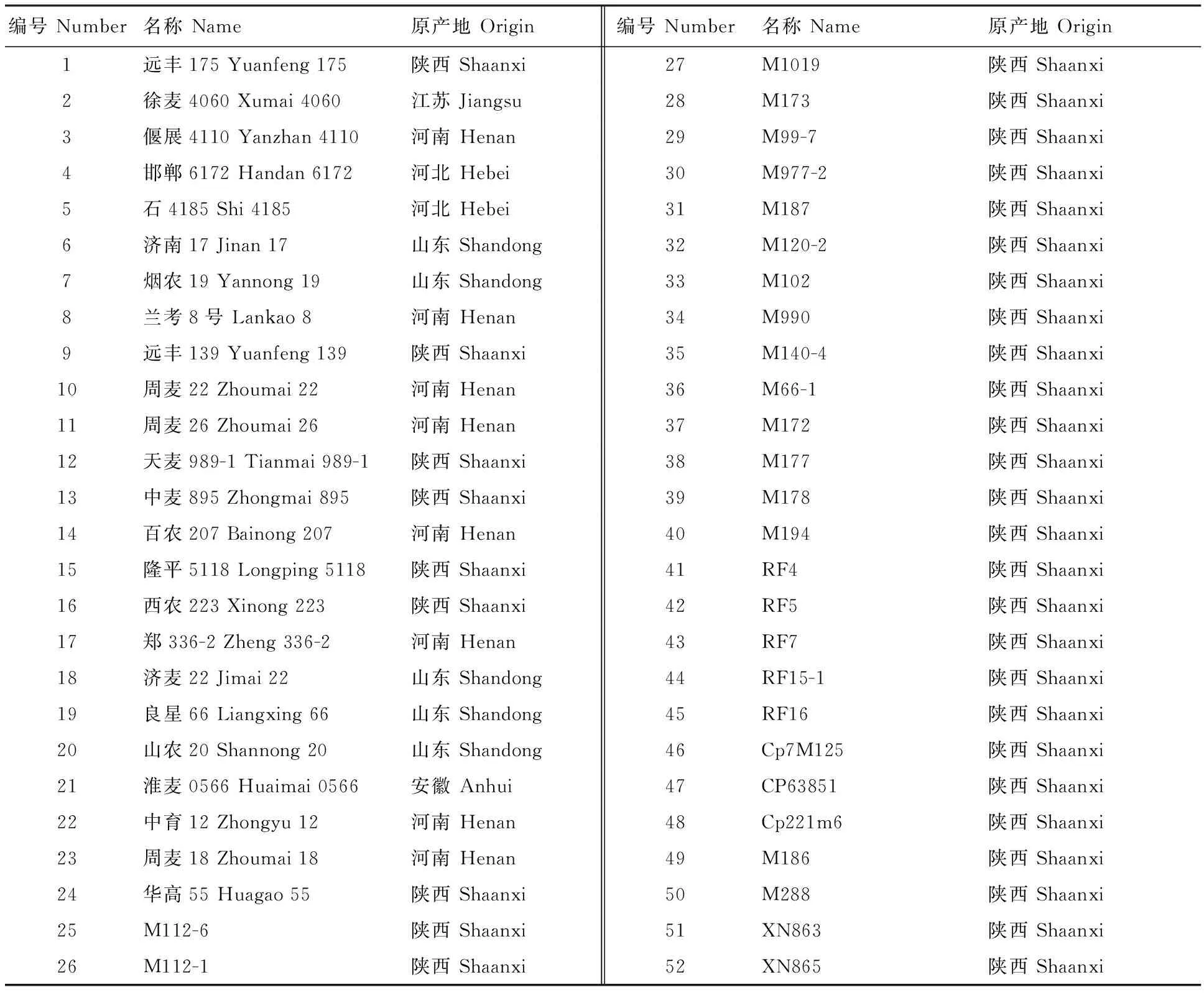
表1 试验材料编号及其来源Table 1 Origin and name of the tested materials
1.4 数据分析
运用SPSS 22.0对表型数据进行描述性统计分析和方差分析,作出5种环境的表型分析表。通过DataFormater软件将,标记的“0,1”带型转为“bp”带型及其他分子遗传学软件可导入的格式。利用Powermarker 进行遗传多样性分析,获得每个分子标记的基因多样性指数、等位变异丰富度及多态性信息含量(polymorphism information content,PIC)等信息。并进行UPGMA聚类分析,结合iTOL(http://itol.embl.de/)网站进行聚类图的绘制与修饰。通过NTsys软件分析获得供试材料的亲缘关系矩阵,并进行亲缘关系聚类热图绘制,并与UPGMA聚类结果进行比对。运用Structure 软件估计52份材料的群体结构,基于贝叶斯方法得到每个材料对应的Q值,将52份材料划分为不同的亚群,绘制群体遗传结构图。
1.5 全基因组关联分析及优异等位变异的发掘
基于供试材料表型数据分析与分子标记遗传多样性、聚类、亲缘关系及群体结构分析,利用获得的表型数据、群体Q值、亲缘关系Kinship值、标记带型等与TASSEL 3.0 软件结合其混合线性模型(multilevel linear model, MLM)进行表型性状与标记间的关联分析。通过关联分析获得关联位点,进行优异等位基因的发掘并计算在P<1/N时对标记位点表型变异的解释率(R2)。
2 结果与分析
2.1 表型统计结果分析
描述性统计分析(表2)表明,不同地区间、性状间小麦两年表型性状差别均较大。在地区间,产量由河南、安徽、陕西依次递减,6个重要产量性状的这种趋势基本一致;在性状间,穗长和单株穂数的变异系数最大,可育小穗数和穗粒数次之,千粒重和株高的变异系数最小。方差分析(表3)表明,6个性状在不同地区间和材料间均存在显著或极显著差异,说明这些小麦性状受环境和遗传因素影响均较大。
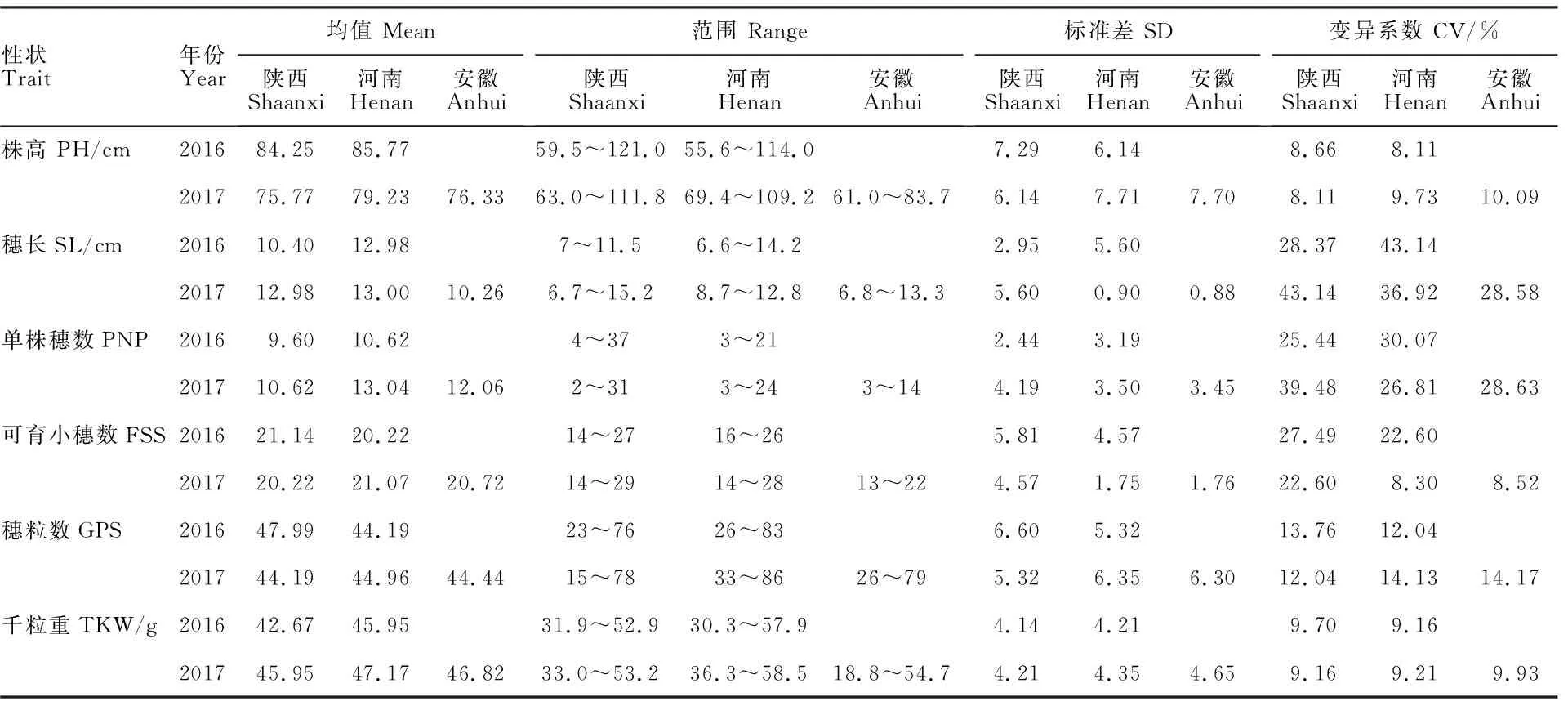
表2 供试群体6个产量性状表型的描述性统计分析Table 2 Descriptive statistics of six yield traits of 52 wheat accessions
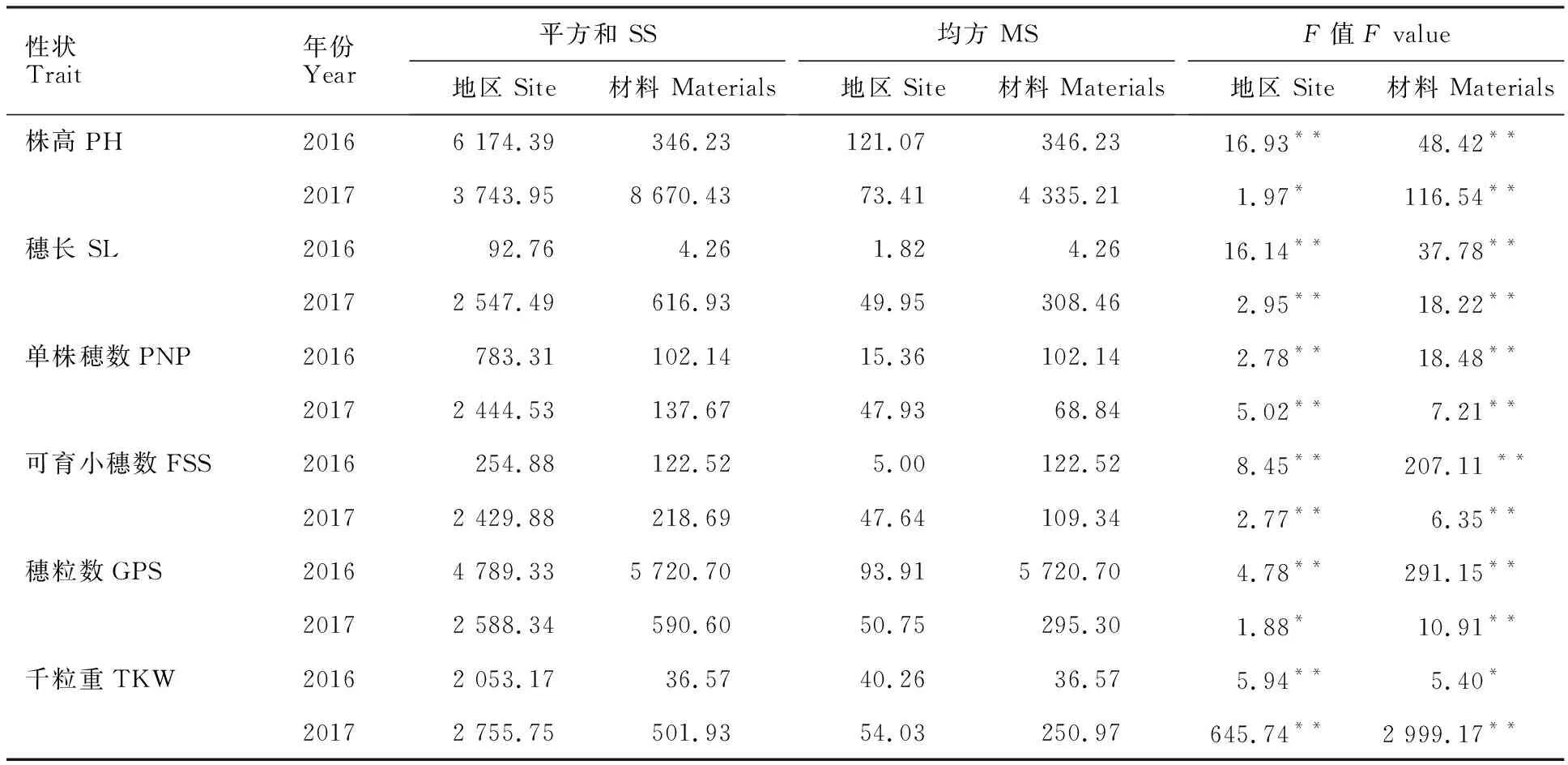
表3 52个小麦品种(系)6个性状两年表型数据方差分析Table 3 ANOVA for six phenotypic traits in 52 wheat varieties(lines) observed in two years
2.2 标记遗传多样性结果分析
利用均匀分布于小麦21条染色体上的248个分子标记,在52份小麦材料中共检测到899个等位变异(表4与图1),每个位点平均检测到3.625个等位变异,变化范围为1~5。遗传多样性指数的变化范围为0.301~0.715,平均值为0.586,其最大和最小值的分子标记分别是gwm635和barc204;多态性信息含量(PIC)变化范围为0.280~0.666,平均值为0.510,达到高度多态的水平(PIC≥0.5),PIC最大和最小值的分子标记分别为gwm635和barc204。在基因组水平上,B染色体组的等位变异总数和平均等位变异丰富度均最高,D染色体组次之,A染色体组最低;A染色体组的遗传多样性指数和多态性信息含量(PIC值)最高,B染色体组中等,D染色体组最低。
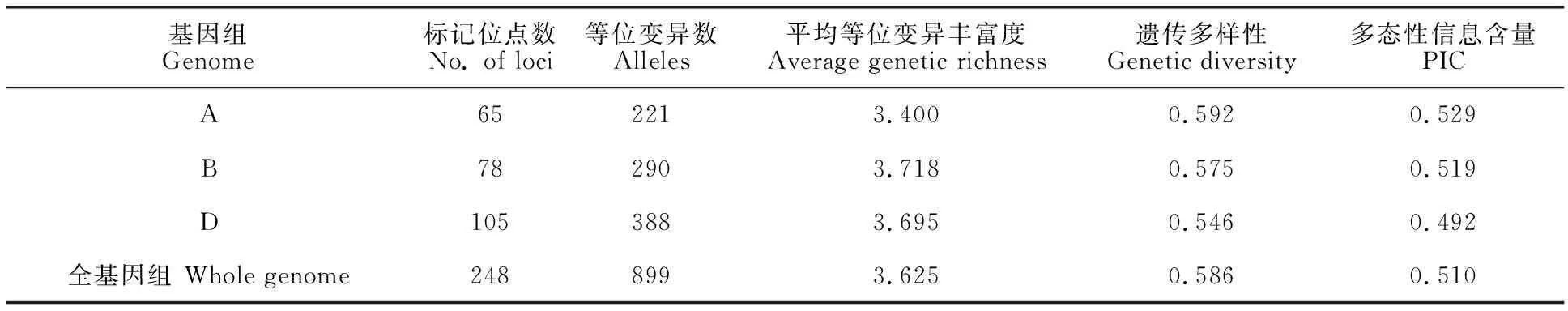
表4 248个标记在供试小麦群体基因组水平上的遗传多样性分析Table 4 Genetic diversity analysis among genome in 52 wheat accessions
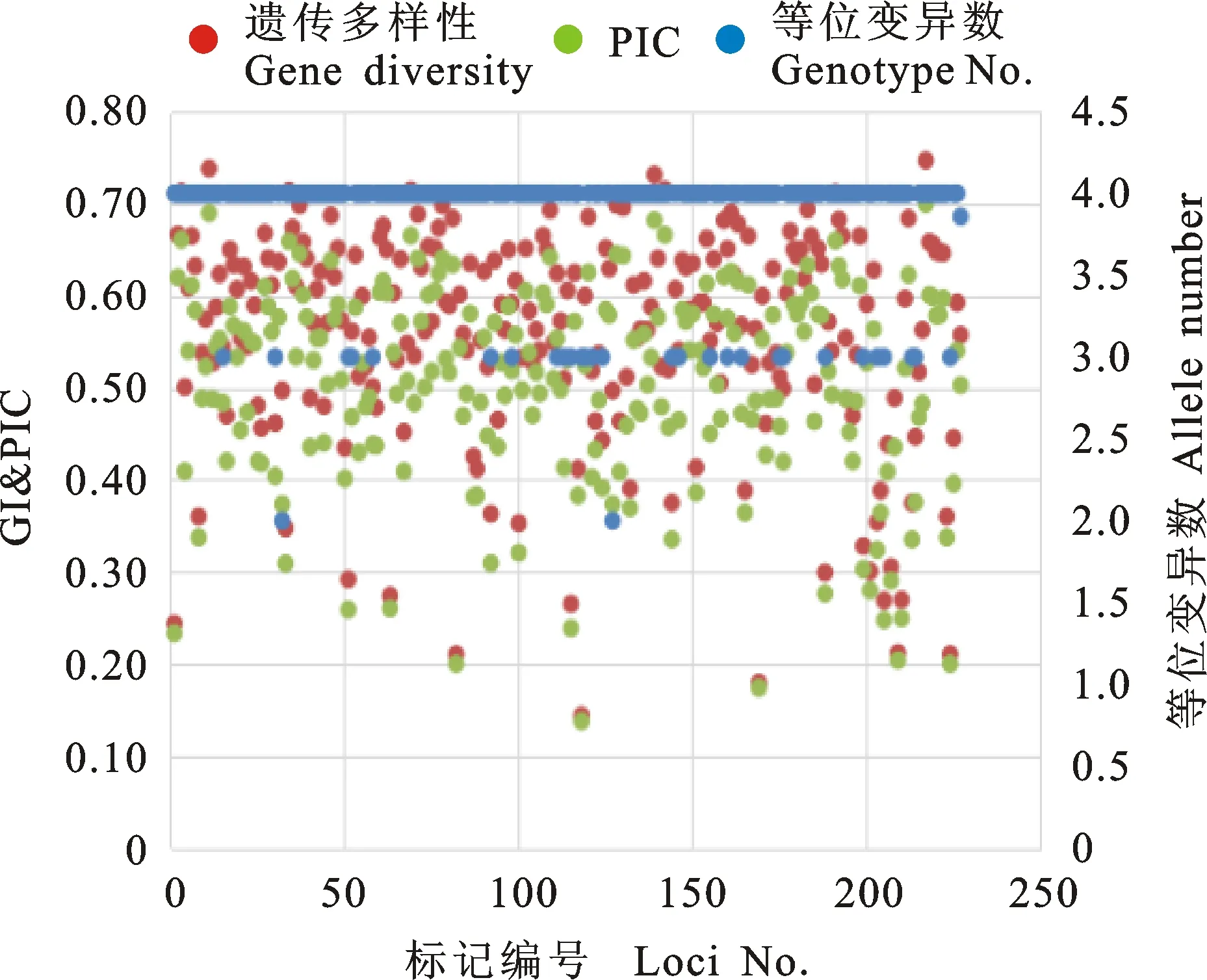
图1 等位变异数与遗传多样性指数(GI)和多态性信息含量(PIC)的散点图Fig.1 Scatter plot of allele number,gene diversity(GI) and PIC
2.3 供试材料群体结构结果分析
利用Structure 2.3.4软件分析,得到52份小麦材料群体结构Q值与极大似然值LnP(D),由LnP(D)计算不同K值间的ΔK来确定最佳K值(图2)。当K=4时,LnP(D)改变平滑趋势且ΔK曲线具有明显的拐点,即52个材料被划分为4个亚群(图3),4个亚群所含品种数分别为12、24、7、9,占总材料数的比例分别为23.1%、46.1%、13.5%和17.3%。分群结果表明,各类群中材料与品种(系)育成时间及地理来源不完全相关。
通过比较UPGMA聚类结果(图4)与利用NTsys得到的亲缘关系(图5),所得群体遗传结构分类结果基本一致,也将群体基本划分为4个亚群,2种分类方法所划分的亚群中,仅编号为20、32、33、42的4个品种(系)分群有差异。
2.4 全基因组关联定位
经关联分析,在两年多点共5种环境下,与供试材料的株高、穗长、单株穗数、可育小穗数、穗粒数及千粒重显著关联的位点有13个,表型变异解释率(R2)为6.74%~82.71%(表5)。与株高、穗粒数、千粒重关联的位点各有1个,分别位于3A、4A、7D染色体上;与穗长关联的位点有5个,其中位于A、B染色体组的位点各2个,位于D染色体组的位点1个;与单株穂数关联的位点有5个,其中位于A染色体组3个,位于D染色体组的位点有2个;与可育小穗数关联的分子标记有3个,其中位于B染色体组的标记2个,D染色体组的标记1个。其中分子标记xwmc580、xwmc181与穗长、单株穗数均显著关联,贡献率最高的分子标记为wmc331。与产量性状显著关联的SNP标记及其引物序列已列出以供参考(表6)。
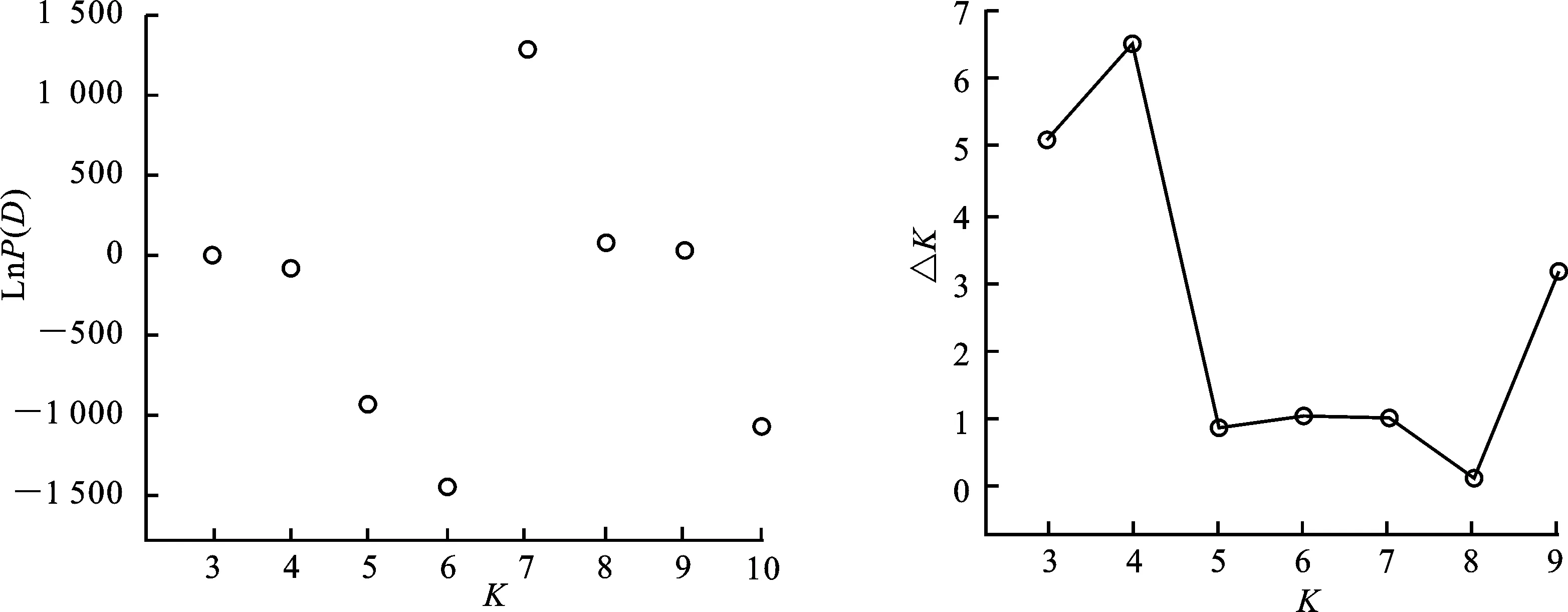
图2 LnP(D)、ΔK估算群体结构图Fig.2 Estimation of population structure by the likelihood distribution and delta K values(ΔK)
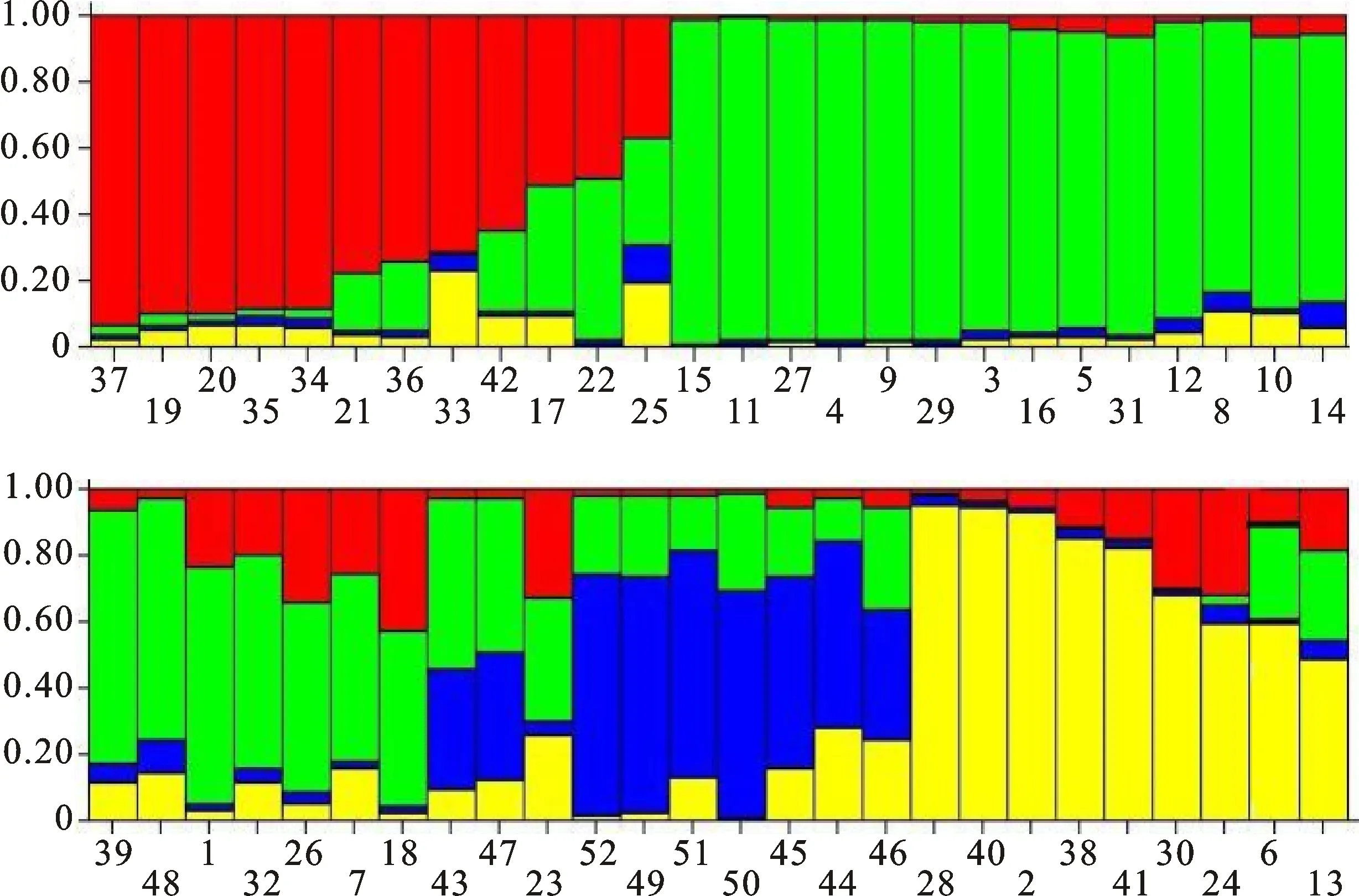
图3 利用 Structure 软件对供试材料的群体遗传结构Fig.3 Population structure(K=4) of materials by Structure software
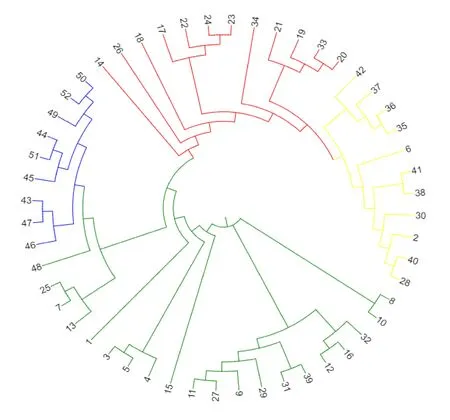
图4 52份供试小麦材料聚类图Fig.4 Dendrogram of 52 wheat accessions by UPGMA cluster analysis
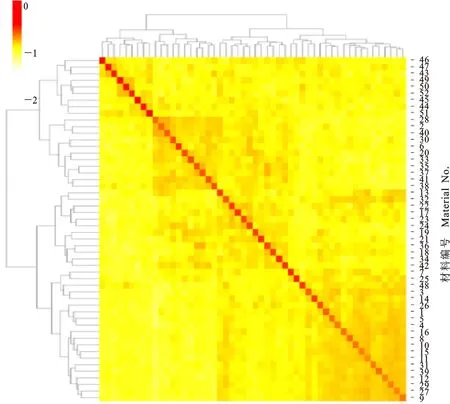
图5 52份供试小麦材料亲缘关系聚类热图Fig.5 Image clustering of 52 wheat accessions by Kingship analysis
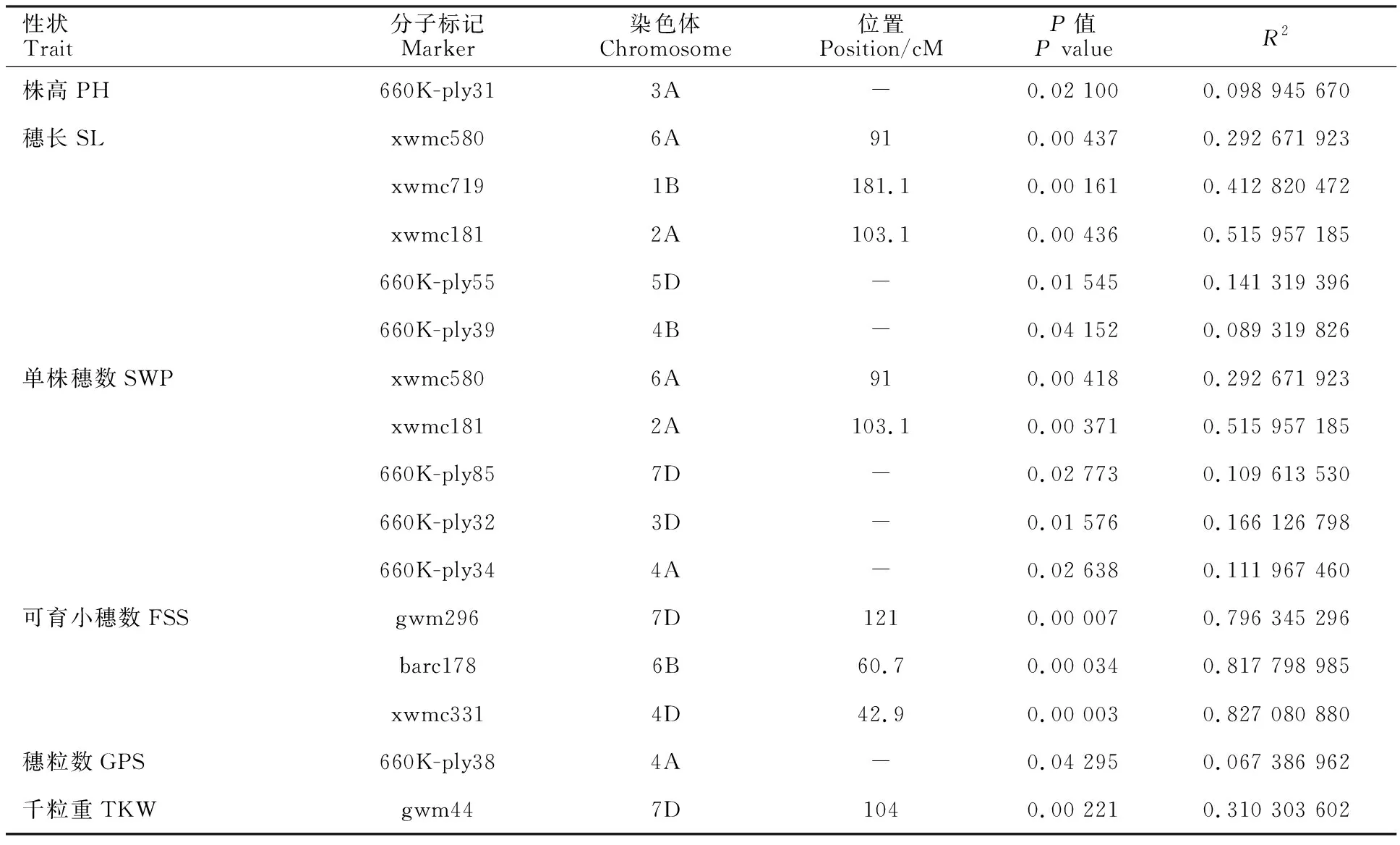
表5 与产量性状显著相关的标记位点Table 5 SSR loci significantly associated with yield-related traits in wheat
表6 所得显著关联SNP标记及其引物序列Table 6 SNP loci and primer sequences significantly associated with yield-related traits in wheat
3 讨 论
在遗传多样性评价体系中,平均等位变异丰富度、基因遗传多样性、多态性信息含量均被普遍应用[20]。本试验所选分子标记平均等位变异丰富度分布(B>D>A)和遗传多样性指数与多态性信息含量分布(A>B>D)不同,且与人们普遍认为的小麦基因组遗传多样性B>A>D有所差异。究其原因,一方面可能由于分子标记只是一段较短DNA序列,不能代表所有基因在染色体上的分布特征,当所用标记数量不同时,分析结果可能不同[21]。如李 远等[22]用86对与赵 檀等[21]用231对多态性SSR引物对相同小麦材料遗传多样性的分析结果差异明显。另一方面,本研究的试验群体较小,且多为陕西省培育的小麦品种(系),可能存在部分基因座,尽管等位变异较少,但其PIC值仍然很高,即引物的多态性信息含量高,其等位变异位点不一定多。赵 檀等[21]、ZHANG等[23]研究认为,评价种质资源遗传多样性不能仅仅用平均等位变异丰富度或基因多样性、多态性信息含量作为评判标准,而有必要对各指标进行权衡。 因此,本研究初步认为,由 52个黄淮麦区小麦品种(系)组成的试验群体,其遗传多样性水平在三个染色体组间没有明显差异。
本试验将52份供试材料分为4个亚群,此分群结果表明各类群中材料与品种(系)育成时间及地理来源不完全相关,分析其原因可能是黄淮麦区生态条件较为相似,育种材料共享力度加大,常用骨干亲本有限,导致了黄淮麦区品种(系)间亲缘关系较近,遗传多样性降低。今后,育种工作应重视扩大引进与利用国外资源,挖掘中国丰富的地方种质资源,不断拓宽改造中国小麦的遗传基础。
本研究中与穗长、单株穗数均显著相关的标记为xwmc181、xwmc580。要燕杰等[24]发现,xwmc580与8个品质和产量性状在0.01水平上显著相关,且其被推测与较好综合农艺性状[25]有关联,在长穗偃麦草研究及小麦遗传改良中有重要利用价值,这与本研究结果一致。xwmc181被定位到2DL,且与小麦黄花叶病毒的抗性相关[26]。此标记也与直立和下披旗叶性状连锁[27]。本试验中与穗长显著相关的标记还有xwmc719,其与水稻植株的矮化性状关联[28],也与小麦抗条锈病性状[29-30]紧密连锁。gwm296、barc178和xwmc331与可育小穗数显著相关。在这三个分子标记中,barc178与小麦花前持续时间呈正相关[31],结合本试验检测结果,推测该区段基因可能与小麦开花期发育相关;gwm296不仅与雌性不育小麦XND126育性基因连锁[32],还与植株的抗叶锈病基因 Lr22a>紧密连锁[33],与抗叶锈病基因 LrSV1>关联[34];xwmc331被定位于4D染色体42.9 cM处[35],其与耐铝[36]、抗赤霉病[37]等性状相关联,是用于小麦育种辅助选择优良分子标记。与千粒重显著相关标记gwm44位于7DS上,是识别小麦持久抗条锈病基因 Yr18>关联的候选标记[38]。综上所述,本试验中与产量性状显著关联位点与前人利用 SSR、QTL等定位位点的位置相同或接近,但所关联性状有所不同。
作为第三代分子标记,SNPs的缺失率明显低于SSRs,SNP具有稳定遗传、高效、快捷的特点[39]。本试验综合两者,用226个微卫星标记与22个SNP标记进行遗传多样性及关联分析,其结果更为可信。已有研究表明,在关联分析中同时使用Q值(群体结构信息)和Kinship值(品种间亲缘关系)的MLM(Q+K)模型优于单独使用Q值或Kinship值的GLM(Q)和MLM(K)模型[40-41]。 为了提高所关联标记的可靠性,本研究对基因型数据采用了Tassel软件中的混合线性模型MLM(Q+K)进行群体结构的分析,最终获得显著关联的13个分子标记,这些优异等位基因有待进一步的精确定位,可为后续相关性状基因的关联定位提供依据。
猜你喜欢
军事文摘(2022年16期)2022-08-24
河南农业·综合版(2022年2期)2022-03-18
河北果树(2021年4期)2021-12-02
今日农业(2021年11期)2021-08-13
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年4期)2020-12-09
医药前沿(2020年20期)2020-11-10
中西医结合肝病杂志(2020年2期)2020-10-27
山西中医药大学学报(2020年2期)2020-06-06
河北农业科学(2019年6期)2019-03-21
