
高效液相色谱-串联质谱法快速测定乌鳢肌肉中金刚烷胺、金刚乙胺和美金刚残留
2018-11-23 03:59李诗言王扬周凡柯庆青贝亦江王鼎南郑重莺吴洪喜丁雪燕
中国渔业质量与标准 2018年5期
李诗言,王扬,周凡,柯庆青,贝亦江,王鼎南,郑重莺,吴洪喜,丁雪燕
(浙江省水产质量检测中心,杭州 310023)
金刚烷胺类药物是一种常见的抗病毒类药物,具有三环胺结构,被广泛应用于对抗流感病毒感染[1-2],主要包括金刚烷胺、金刚乙胺和美金刚(化学结构式见图1)。由于在畜牧业滥用金刚烷胺类药物会带来耐药性及食物链传递导致的人体过量摄入等问题,中国农业部早在2005年就已经明确禁止其在动物养殖业中使用[3]。乌鳢(OphiocephalusargusCantor)肉质细嫩,是优质的水产养殖品种之一,长期以来乌鳢的养殖摄食以冰鲜杂鱼为主,但近两年冰鲜鱼价格上涨而鸡肝、鸭肝却有着较充足的供应量,因此不少养殖户纷纷转用鸡肝、鸭肝饲养乌鳢[4]。然而,少数不法分子仍然会在畜禽养殖过程中使用金刚烷胺类药物进行禽流感的预防和早期治疗[5-7]。通过食物链的传递,鸡肝、鸭肝中存在的金刚烷胺类药物就可能在乌鳢体内蓄积,从而产生新的水产品质量安全问题,因此非常有必要建立乌鳢肌肉中高效、准确、灵敏的金刚烷胺类检测分析方法。
近年来,生物基质中金刚烷胺类药物的检测方法已在文献中广泛报道,包括气相色谱法[8]、气相色谱-串联质谱法[9]和液相色谱-串联质谱法(LC-MS/MS)[10-18],其中LC-MS/MS方法因具有高特异性,高灵敏度且无需衍生化,应用范围最为广泛。目前金刚烷胺类药物检测方法大都针对鸡肉、鸡蛋等禽类样品,在水产品中的检测方法鲜有报道。通常金刚烷胺类药物主要通过C18色谱柱[10-15]或者亲水作用(HILIC)色谱柱[16-18]进行分析,但是金刚乙胺和美金刚互为同分异构体,在LC-MS/MS分析中对色谱分离上有着更严苛的要求。本研究在样品前处理上采用通过式固相萃取技术,所用试剂少、效率高,比较了不同类型色谱柱对金刚烷胺、金刚乙胺和美金刚的分离效果,并在此基础上建立了优化的LC-MS/MS分析条件,实现了乌鳢肌肉中3种金刚烷胺类药物的快速准确分析。
1 材料与方法
1.1 仪器与试剂
实验仪器主要包括ExionLC AD系列液相色谱仪(美国 SCIEX公司)、4500 QTRAP 三重四级杆质谱仪(美国 SCIEX 公司)、Multi Reax全能型振荡器(德国 Heidolph 公司)、Allegra X-12高速离心机(美国 BECKMAN 公司)和N-EVAP112水浴式氮吹浓缩仪(美国Organomation Associates,Inc)。
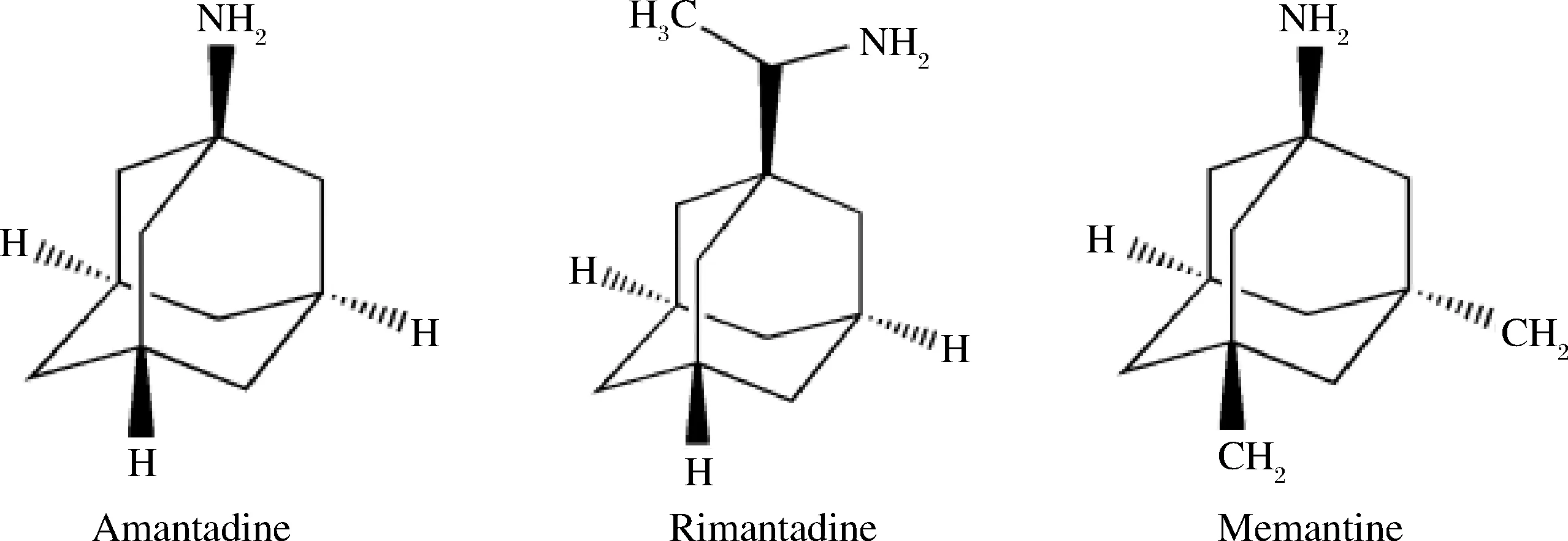
图1 金刚烷胺、金刚乙胺和美金刚的化学结构图Fig.1 Chemical structures of amantadine,rimantadine and memantine
实验中用到的标准物质包括金刚烷胺(amantadine, AT),金刚乙胺(rimantadine-D6, RT),美金刚(memantine, MT),金刚烷胺-D6(amantadine D6, AT-D6)和美金刚-D6(memantine-D6, MT-D6),5种标准物质的纯度均大于95%,由加拿大TRC公司生产。实验中用到的主要耗材及试剂包括Oasis PRiME HLB固相萃取柱(200 mg/6 mL,美国Waters公司),乙腈、甲醇(色谱纯,美国TEDIA公司),甲酸(色谱纯,美国ROE Scientific INC.)和乙酸铵(色谱纯,美国Fisher 公司)。
1.2 标准溶液的配制
分别称取金刚烷胺、金刚乙胺、美金刚和2种同位素内标各10 mg,以甲醇为溶剂分别定容于25 mL容量瓶中,配制成质量浓度为400 mg/L的标准物质储备液,-20 ℃储存。用上述储备液准确配制质量浓度为1 mg/L的内标混合工作液以及质量浓度为0.1mg/L的混合标准工作液,-20 ℃储存。
1.3 样品制备
样品采自浙江省内乌鳢养殖场,分别取肌肉组织,清水洗净后用干纱布小心吸除多余水分,放入搅拌机中制为匀浆状,置于-20 ℃冻藏备用。
1.4 样品前处理
1.4.1 样品提取
准确称取乌鳢肌肉2.00 g,置于50 mL带盖的离心管中,加入50 μL内标混合工作液,移取5 mL 80%(V/V)的乙腈水溶液(含0.1%甲酸,V/V)于离心管中,充分涡旋混匀1 min后超声提取5 min,8 000 r/min的速度高速离心5 min,转移全部上清液于10 mL带刻度比色管中,再向离心管中加入4 mL 样品提取液,重复上述提取步骤,合并提取液,并用80%(V/V)的乙腈水溶液(含0.1%甲酸,V/V)定容至10 mL,待净化。
1.4.2 样品净化
准确移取2.5 mL 上述带基质样品提取液,加于Oasis PRiME HLB柱(200 mg/6 mL)上,控制流速为1滴/s,自然流干后再加入1.0 mL 80%(V/V)乙腈水溶液(含0.1%甲酸,V/V)润洗柱子,收集全部流出液,于40 ℃下氮吹至近干。用0.5 mL 20%(V/V)乙腈水溶液复溶,溶液过0.22 μm有机相滤膜,上机检测。
1.5 LC-MS/MS条件
1.5.1 色谱条件
色谱柱为Phenomenex Kinetex F5(2.1 mm×100 mm,2.6 μm);流动相A为乙腈,流动相B为5 mmol/L乙酸铵水溶液(含0.1%甲酸,V/V);柱温为40 ℃;流速为0.4 mL/min;进样量为2 μL。梯度洗脱程序:0~1.0 min,97% B;1.0~1.1 min,97%~85% B;1.1~3.6 min,85%~65% B;3.6~5.6 min,65%~10% B;5.6~6.6min,10%B;6.6~6.7min,10%~97% B;6.7~9.0min,97% B。
1.5.2 质谱条件
离子源为电喷雾离子源,正源模式,离子源温度550 ℃,电喷雾电压5 500 V,雾化气(GS1)压力345 kPa,辅助气(GS2)压力379 kPa,气帘气(CUR)压力172 kPa,扫描方式为多反应监测(MRM)模式,具体质谱参数见表1。
2 结果与讨论
2.1 质谱条件优化
将目标药物用甲醇稀释配置成质量浓度为0.5 mg/L的上机工作液,采用针泵进样进行质谱条件优化。由于金刚烷胺、金刚乙胺和美金刚均含有氨基,因此在ESI+模式下响应较好,主要形成准分子离子峰[M+H]+。在确定目标化合物母离子后进一步对其进行子离子扫描,其中金刚乙胺和美金刚互为同分异构体(图1),二者子离子碎片具有高度的一致性,但相对丰度比却存在着差异(图2)。本研究选择相对丰度最强的180.1>163.1同时作为金刚乙胺和美金刚的定量离子对,180.1>107.1作为美金刚的定性离子对,180.1>81.1作为金刚乙胺的定性离子对,这与Mu等[13]和Yumi等[15]的研究结果一致。优化后3种金刚烷胺类药物及2种同位素内标物的MRM质谱参数见表1。
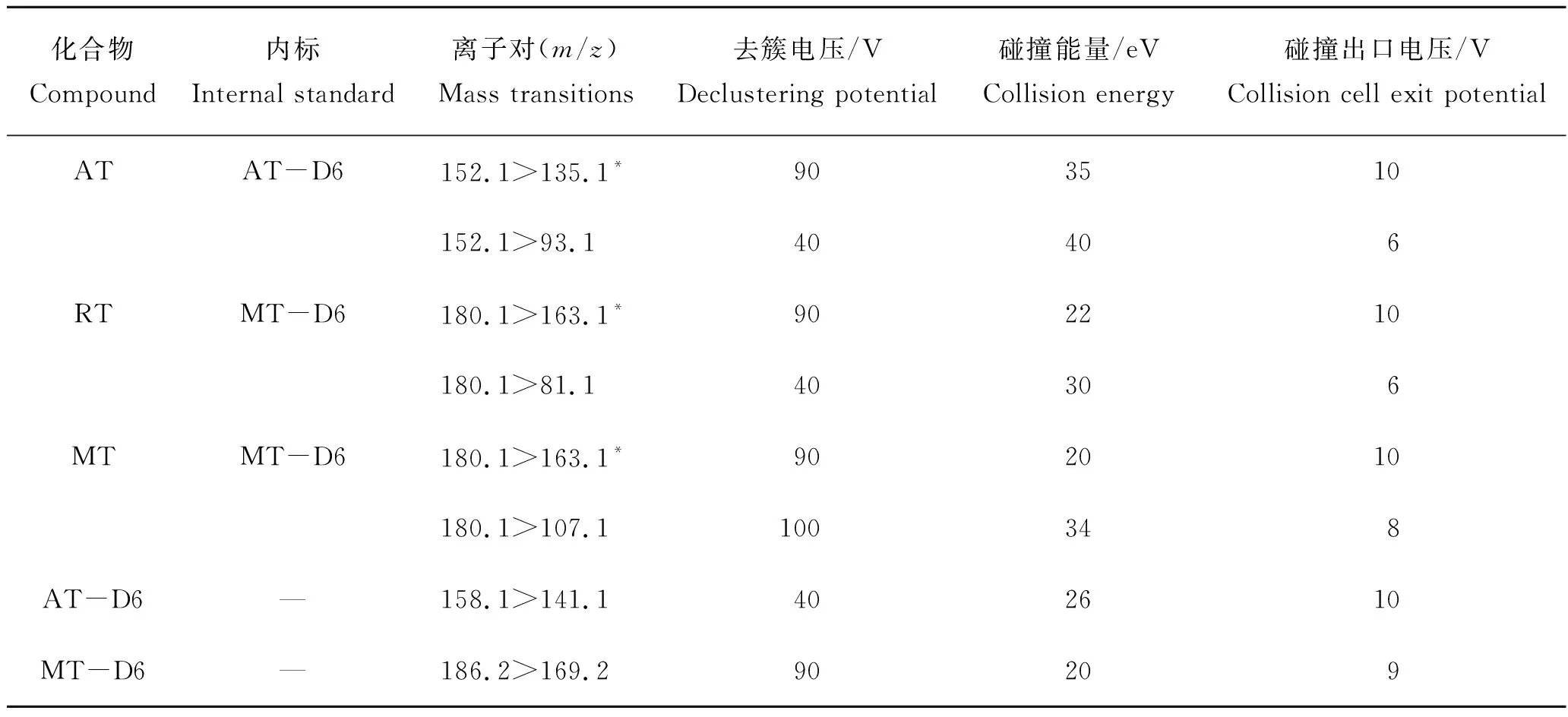
表1 目标物化合物的质谱参数Tab.1 Mass spectrometric parameters for target compounds
注:*表示定量离子。“—”示无。AT为金刚烷胺(amantadine),RT为金刚乙胺(rimantadine),MT为美金刚(memantine),AT-D6为金刚烷胺-D6(amantadine-D6),MT-D6为美金刚-D6(memantine-D6),下同。
2.2 色谱条件的选择
金刚乙胺和美金刚在质谱条件下无法完全区分,因此选择合适的色谱条件对两者进行分离是本研究的难点之一。实验考察了4种型号的色谱柱对目标药物的分离情况,分别是Waters ACQUITY BEH Amide(2.1 mm×100 mm,1.7 μm)、Thermo Fisher Syncronis HILIC(2.1 mm×100 mm,1.7 μm)、Waters ACQUITY BEH C18(2.1 mm×100 mm,1.7 μm)和Phenomenex Kinetex F5(2.1 mm×100 mm,2.6 μm),并针对不同类型色谱柱的流动相进行了选择和优化。结果可知BEH Amide和Syncronis HILIC色谱柱在HILIC分离模式下难以完全分离金刚乙胺和美金刚,方法适用性较差(图3-1和图3-2),BEH C18色谱柱使用甲醇能够基本实现金刚乙胺和美金刚的分离(图3-3),而乙腈由于洗脱能力更强不能对两者实现分离,这与相关研究报道[10-11]的结果一致;Kinetex F5色谱柱使用甲醇后金刚乙胺和美金刚虽能分离,但峰形较差,而乙腈可使两者色谱峰峰形更加对称尖锐,同时实现基线分离。从图3-3和图3-4的对比可以看出,Kinetex F5色谱柱对金刚乙胺和美金刚的分离情况要好于BEH C18色谱柱,其填料含有五氟苯基,可与化合物发生偶极-偶极、电荷转移和离子交换等相互作用,提高了对异构体化合物的分离度[19],是本方法中最为合适的分析柱。此时,液相方法流动相有机相为乙腈,而水相为5 mmol/L乙酸铵水溶液(含0.1%甲酸,V/V),具体优化后的色谱条件见1.5.1节。
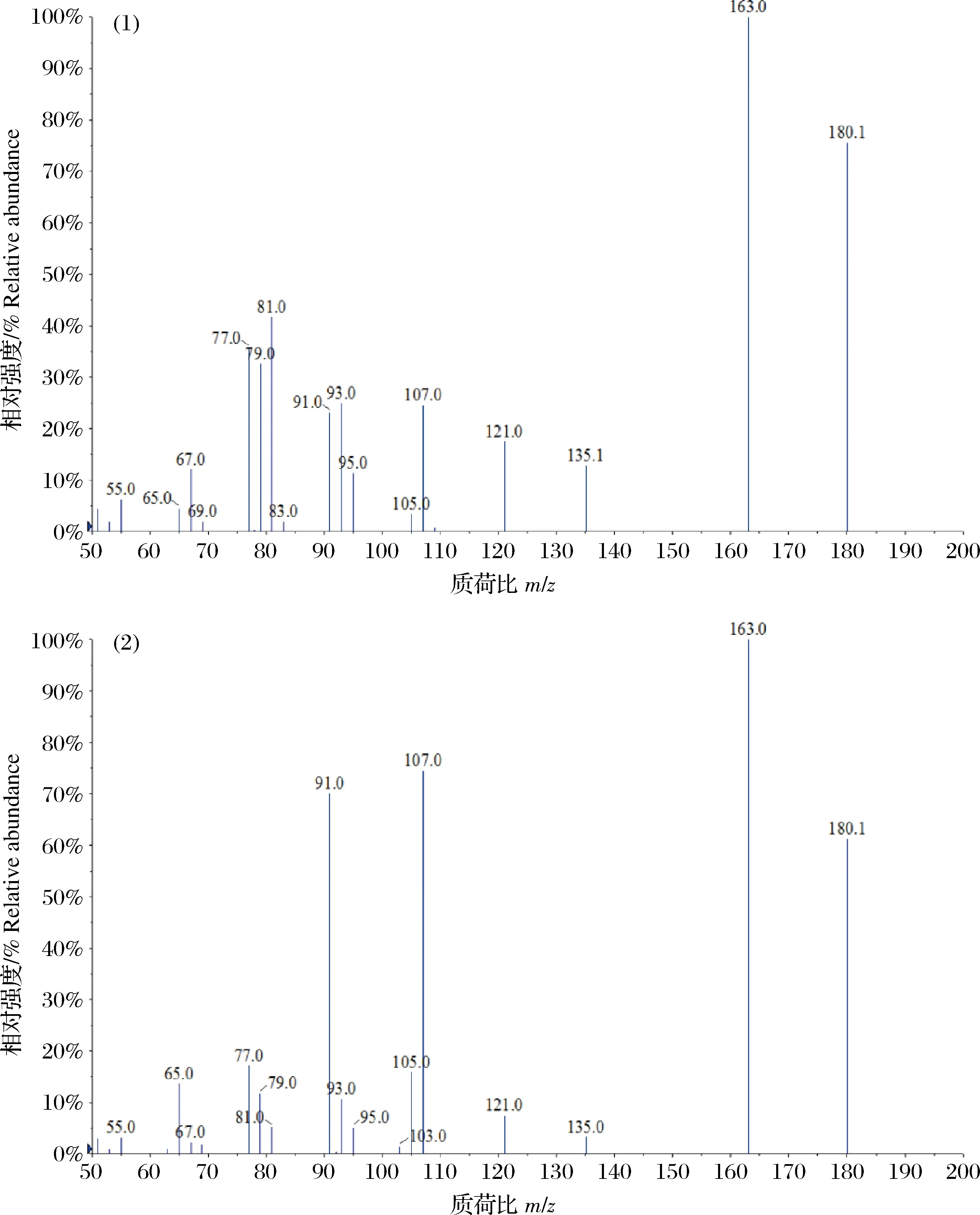
图2 金刚乙胺和美金刚的子离子质谱图(1)金刚乙胺;(2)美金刚。Fig.2 The daughter ion spectrums of rimantadine and memantine(1)Rimantadine;(2)Memantine.
2.3 样品前处理的优化
金刚烷胺类药物均含有氨基,属于碱性化合物,在酸性溶液中易溶于极性试剂,通常其提取试剂主要为乙腈或甲醇与酸性溶液配制而成的混合提取液[10,11,13-18]。本研究中乌鳢样品的主要基质干扰成分为蛋白和脂类物质,采用乙腈作为提取试剂具有良好蛋白沉淀效果,而PRiME HLB小柱能够去除样品基质中的磷脂等非极性物质。参考文献[15],本研究最终采用乙腈水溶液(含0.1%甲酸,V/V)作为样品提取液,同时使用PRiME HLB通过式固相萃取小柱对样品进行净化,省去了常规固相萃取方法的淋洗和洗脱步骤,具有高效和高通量的特点[20]。
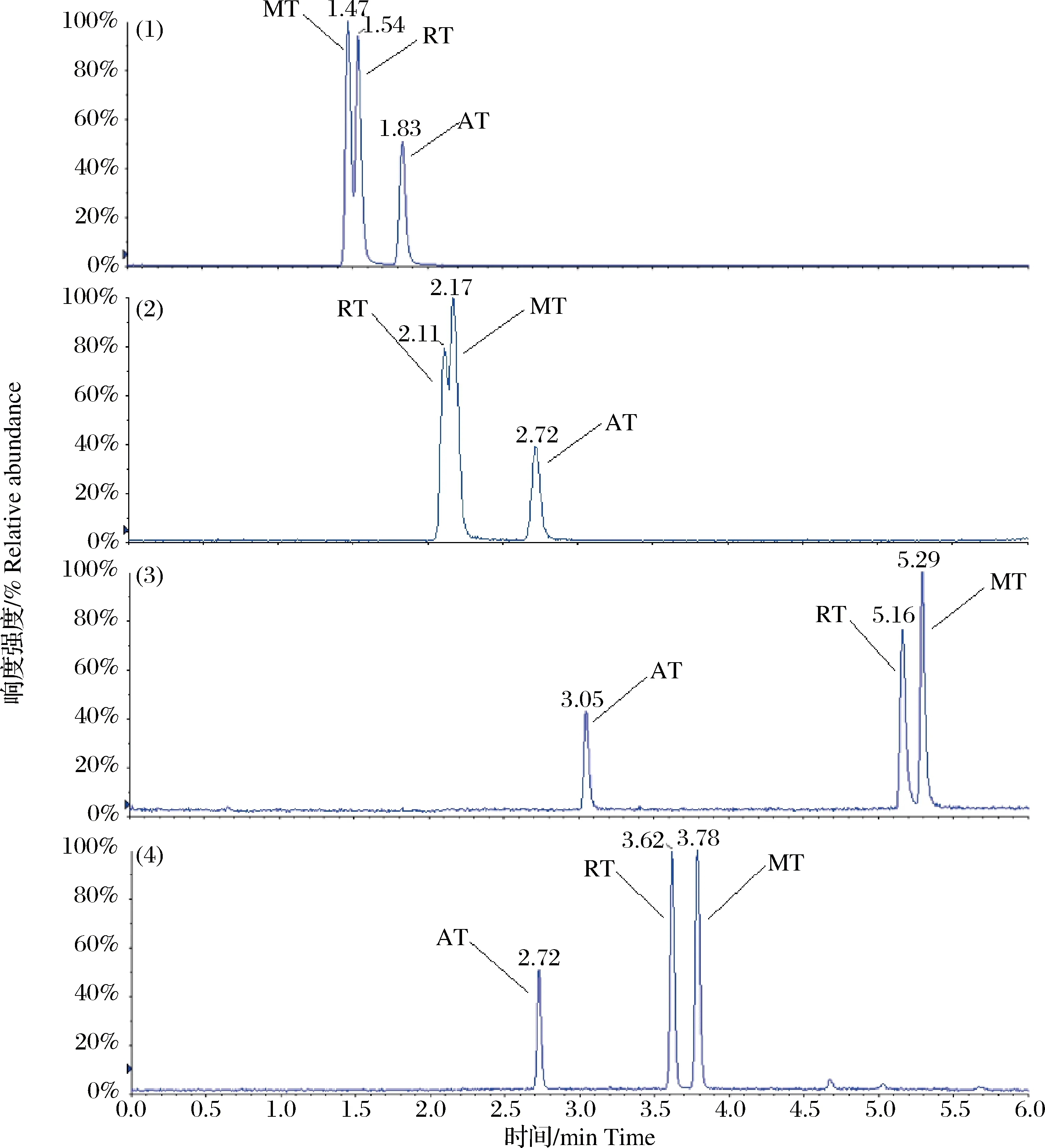
图3 不同色谱柱分离条件下目标化合物标准溶液(2 μg·L-1)的液相色谱-串联质谱总离子流图(1)BEH Amide色谱柱;(2)Syncroni HILIC色谱柱;(3)BEH C18色谱柱;(4)Kinetex F5色谱柱。 AT: 金刚烷胺;RT: 金刚乙胺;MT: 美金刚,下同。Fig.3 The total ion chromatograms (TICs)of target compounds in standard solutions (2 μg·L-1) by LC-MS/MS analysiswith different chromatographic column(1)BEH Amide;(2)Syncroni HILIC;(3)BEH C18;(4)Kinetex F5. AT: amantadine;RT: rimantadine;MT: memantine. The same below.
提取液中乙腈的比例会直接影响目标化合物的提取效率和净化效果,为此我们考察了不同提取试剂下经PRiME HLB小柱处理后的净化效果。分别向6份空白乌鳢样品中加入适量标准工作液,3种药物的加标水平均为5μg/kg,按照1.4.1节方法获得加标样品提取液共计6份,按照1.4.2节方法取1 mL提取液通过SPE小柱获得净化后基质溶液,以绝对回收率(absolute recovery,AR)考察了不同乙腈比例提取液的方法适用性。AR计算公式如下:AR(%)=(加标样品溶液信号强度/空白试剂加标溶液信号强度)×100%;6份样品的AR数据合并后按“均值±标准差”进行处理。如图4所示,随着乙腈比例的不断提高,3种药物的绝对回收率均为先快速升高再缓慢下降的趋势。进一步实验表明,当乙腈比例低于70%时目标化合物在SPE柱上无法完全被洗脱下来,影响了绝对回收率。当乙腈比例高于70%时,随着乙腈比例的进一步提高,提取液极性变弱且洗脱能力变强,这会导致目标化合物提取率的逐步下降并减弱了SPE小柱的净化效果,造成基质效应的增加。由于金刚烷胺和金刚乙胺基质效应为增强效果,部分抵消了提取率的下降值,导致两者绝对回收率下降幅度较低,而美金刚基质效应为抑制效果,因此绝对回收率下降幅度较大。最终,本研究选用乙腈比例为80%的样品提取液,此时金刚烷胺、金刚乙胺和美金刚的绝对平均回收率相对较高,分别为93.7%、87.2%和75.1%,且稳定性较好,具有良好的方法适用性。
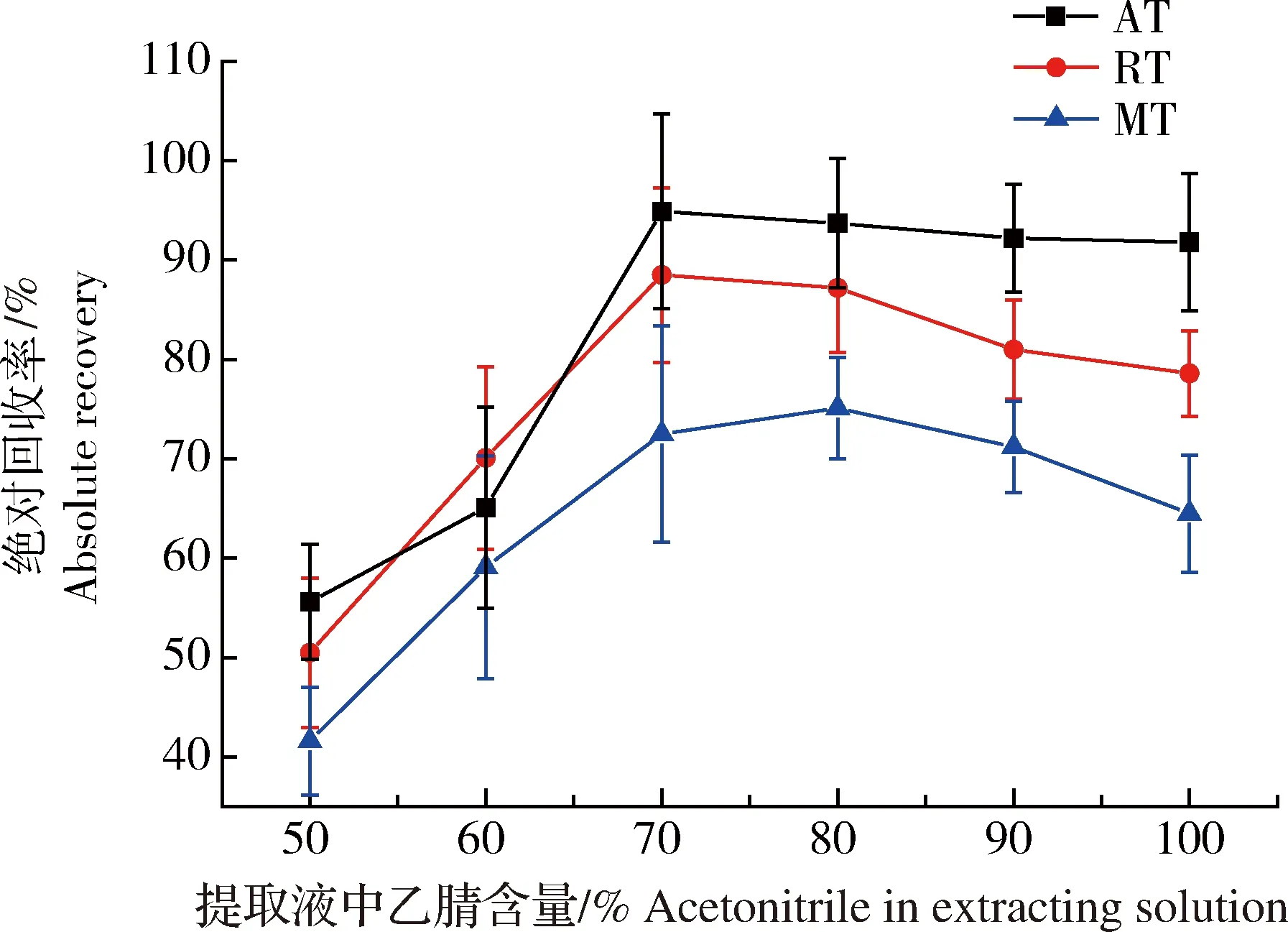
图4 提取液中不同比例乙腈对通过式固相萃取净化后金刚烷胺、金刚乙胺和美金刚绝对回收率的影响(n=6)Fig.4 The effects of different acetonitrile contents (%) in extracting solution for the absolute recovery of amantadine, rimantadine and memantine using an pass-through solid phase extraction purification methods(n=6)
SPE小柱的柱容量有限,上样量过大会影响样品的净化效果,上样量过小则会增大稀释倍数造成方法灵敏度的降低,因此需要确定合适的上样体积。本研究考察了上样量对目标化合物净化效果的影响,发现提取液的上样量超过3 mL后,净化后样品金刚乙胺和美金刚的基质效应开始显著增加,考虑到稀释倍数整除需要,最终确定上样量为2.5mL(相当于0.5 g 鱼肉基质)。
2.4 基质效应的影响
基质效应(matrix effect,ME)在液相色谱串联质谱分析中普遍存在,是影响检测结果准确性的重要因素之一,在质谱检测中共流出干扰物可对目标化合物产生一定程度的离子抑制或者增强作用[19-21]。本研究基质效应计算公式如下:ME(%)=(基质空白溶液加标信号强度/空白溶液加标信号强度)×100%。实验发现金刚烷胺和金刚乙胺存在基质增强作用,美金刚存在基质抑制作用,三者基质效应分别为113%、107%和84%(表2)。为确保检测结果准确性,本研究采用同位素稀释内标法进行定量。
2.5 方法学考察
2.5.1 方法的线性范围、回归方程与定量限
采用标准物质混合工作液和内标配制成6种质量浓度的标准系列梯度,依据优化好的色谱、质谱条件对6个浓度水平的系列梯度工作溶液进行测定。3种目标化合物均以其定量离子对峰面积与相应同位素内标峰面积的比值(y)为纵坐标,以质量浓度(x,μg/L)为横坐标做内标定量工作曲线。结果见表2,3种目标物的线性范围均为0.5~100.0 μg/L,相关系数(r2)为0. 999 3~0. 999 7,表明各化合物具有良好的线性关系。采用标准添加法进行定量限(LOQ)和检出限(LOD)验证,以信噪比S/N大于3为方法的LOD,同时LOQ须满足信噪比S/N大于10。本方法中金刚烷胺、金刚乙胺和美金刚的LOD值分别为0.2、0.1和0.1 μg/kg;在0.5 μg/kg水平标准添加时,上述3种目标化合物的信噪比S/N分别为17、35和42,满足作为定量限的要求。本方法中,金刚烷胺、金刚乙胺和美金刚的LOQ均为0.5 μg/kg,优于标准SN/T 4253—2015[16]的1 μg/kg。
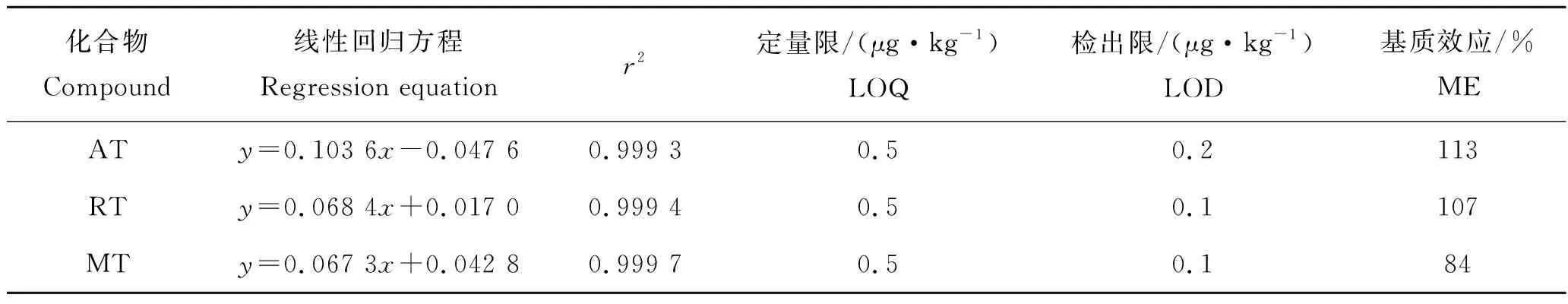
表2 3种目标化合物的回归方程、相关系数、检出限和定量限Tab.2 Regression equations and correlation coefficients (r2) of three analytes
2.5.2 回收率和精密度
按前述方法,取空白乌鳢肌肉样品,做3个添加水平的加标回收试验,每个添加水平做6次平行测定,结果见表3。方法中3种目标物的平均回收率为91.7%~104.0%,RSD为4.6%~8.1%,说明方法的精密度和准确度可满足分析检测要求。如图5所示,乌鳢空白加标样品各化合物色谱峰形良好,且目标物出峰处无杂质峰干扰。
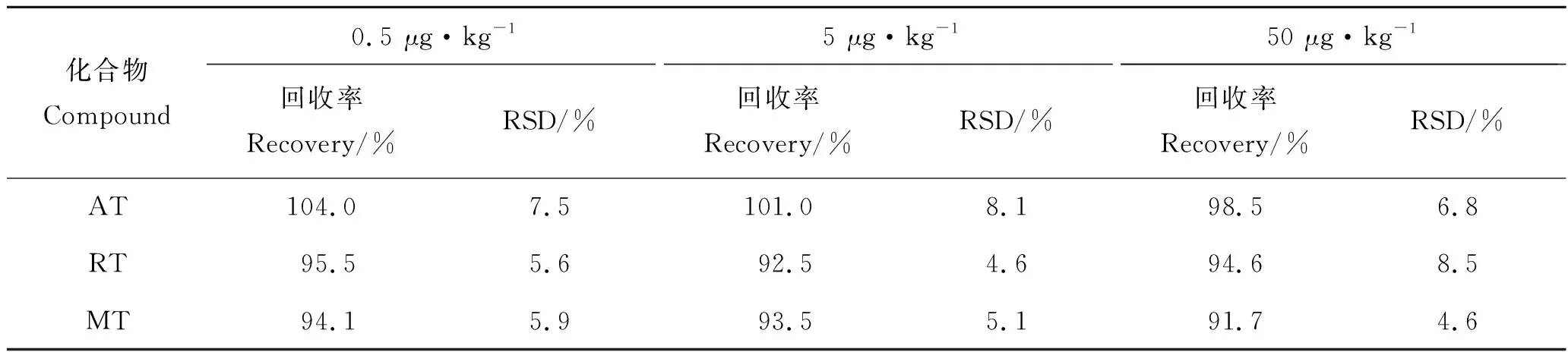
表3 空白乌鳢肌肉中金刚烷胺、金刚乙胺和美金刚的加标回收率和精密度 n=6
2.6 实际样品检测
采用本方法对24批次乌鳢样品进行检测,所有样品均未检出金刚烷胺类药物残留。质控样品金刚烷胺、金刚乙胺和美金刚的加标量均为2.0 μg/kg,平均回收率分别为95.0%、94.5%和96.0%,RSD范围为3.78%~6.71%,同时采用国家标准SN/T 4253—2015[16]对上述样品进行复测,检测结果一致,说明本研究方法可靠有效。
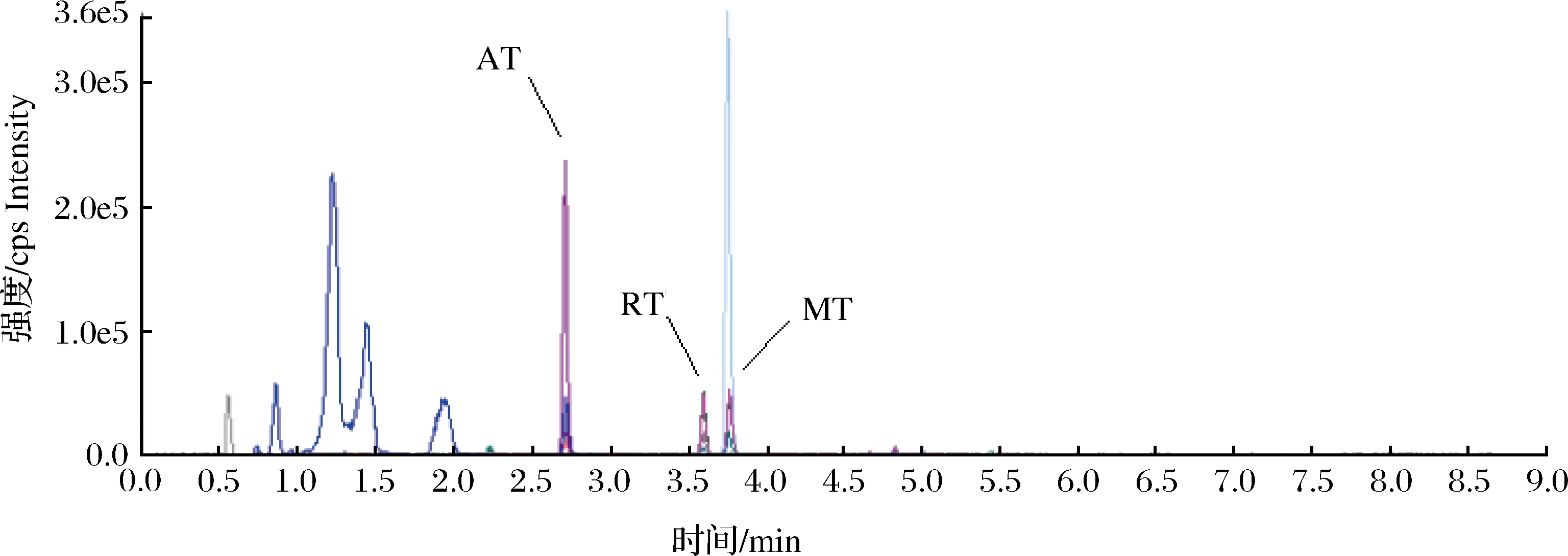
图5 乌鳢加标样品中目标物的MRM图(5 μg·kg-1)Fig.5 MRM chromatograms of blank Ophiocephalus argus Cantor muscle added with the three drugs (5 μg·kg-1)
3 结论
本研究该方法采用基于通过型固相萃取技术的前处理方法,建立了乌鳢肌肉中金刚烷胺、金刚乙胺和美金刚的LC-MS/MS测定方法,采用Kinetex F5色谱柱实现了金刚乙胺和美金刚的有效分离,通过同位素稀释内标法定量完成了24批次养殖乌鳢样品的检测。相比基于经典固相萃取的前处理方法,本方法前处理简便快捷,极大地提高了检测效率,且适用于现有检测标准要求,可用于乌鳢肌肉中3种金刚烷胺类药物的快速筛查与准确定量。
猜你喜欢
电脑迷(2022年3期)2022-11-08
中国农学通报(2022年20期)2022-08-19
西南农业学报(2022年6期)2022-08-06
毛纺科技(2021年8期)2021-10-14
天津药学(2021年1期)2021-03-31
世界科学技术-中医药现代化(2020年2期)2020-07-25
浙江农业科学(2019年7期)2019-07-22
中国粮油学报(2019年4期)2019-07-12
农业知识(2019年5期)2019-03-26
中成药(2017年4期)2017-05-17
