
Genomic heterogeneity meets cellular energetics:crosstalk between the mitochondria and the cell cycle
2018-11-21 01:58:16EricaHerreraSehamAzzamMadisonBergerLauraDiazMartinez
Erica L. Herrera, Seham Z. Azzam, Madison C. Berger, Laura A. Diaz-Martinez
Department of Biological Sciences, The University of Texas at El Paso, El Paso, TX 79968, USA.
Abstract Changes in cellular energetics and genomic instability are two characteristics of cancers that have been studied independently. Evidence of cross-talk between mitochondria function and nuclear function has started to emerge,suggesting that these pathways can influence one another. Here we review recent evidence that links the mitochondria and the cell cycle. This evidence indicates bidirectional cross-talk where mitochondria function can regulate the cell cycle and induce genomic instability, and conversely, the cell cycle machinery regulates mitochondria function.Implications for this cross-talk in the development of cancer are discussed.
Keywords: Mitochondria dynamics, cell cycle, mitochondria heterogeneity, genomic heterogeneity
INTRODUCTION
Changes in metabolism and genomic instability were among the earliest characteristics of tumors to be identified. Boveri’s hypothesis in the early 1900s that malignant tumors originated from cells with abnormal chromosome numbers[1], initiated an era of research on the role of genomic instability in cancer development.Likewise, Otto Warburg’s work on the metabolic changes in tumor cells[2,3]pioneered an era of research studying the role of changes in cell metabolism during cancer progression. These twofields, however, have mostly remained separate. Here we focus on emerging evidence of crosstalk between the processes occurring at the mitochondria and those in the nucleus, particularly as it relates to the cell cycle. These discoveries suggest an important connection between mitochondria heterogeneity and genomic heterogeneity that has key implications for our understanding of cancer development.
GENOMIC INSTABILITY IN CANCERS
Genomic instability has long been associated with cancer development[4]and can range from single point mutations[5,6]to massive genomic rearrangements (e.g., chromothripsis)[7,8]. Parallel genome sequencing studies of cancer cells have revealed a wide variety of mutations and chromosomal abnormalities existent in cancer genomes: studies looking at mutations identified averages of 47-84 non-silent clonal mutations per tumor[9].Studies focusing on clonal somatic chromosomal rearrangements have uncovered similar variation, from cancer cells containing a single chromosomal rearrangement per cell to cells with > 200 rearrangements, including deletions, duplications, and inversions[10,11]. Similarly, studies looking at gene copy number abnormalities identified a mean of 209 somatic copy number abnormalities per cancer genome[12]. A commonfinding throughout these studies is the great heterogeneity in the type of genomic instability as well as in the identity of the genes affected, with only a small number of genes found to be commonly affected in multiple cancers.The complexity of genomic heterogeneity in cancer is further expanded when we consider that these differences are not limited to differences among clonal populations of cancer cells (inter-tumoral heterogeneity). Sequencing of different regions within a tumor reveals equally staggering intra-tumor genomic heterogeneity[13-17], which is dynamic over time[18-22]. Taken together, these results are consistent with models of rapid genomic evolution within tumors and intra-tumoral genomic heterogeneity increasing over time, correlating with tumor aggressiveness and decreased patient survival[23,24].
Genomic instability can be initiated by exogenous or endogenous agents. The role of external genotoxic agents (e.g., UV-light, x-rays, chemical mutagens) in inducing genomic changes has been extensively reviewed elsewhere[25-28]. Endogenous causes of genomic instability include errors in DNA replication[29], transcriptioninduced stress[30], spontaneous or activation-induced cytosine deamination[31], transposon mobilization[32],and defective or error-prone DNA repair[33], among other factors. Another important and widely studied source of genomic instability is DNA damage induced by the reactive oxygen species (ROS) produced in the mitochondria during the respiration process. ROS function in the cell as signaling molecules that regulate multiple cellular pathways and are key for cell and organism homeostasis[34]. However, ROS can also generate direct DNA damage by oxidation of DNA bases[35,36]. Importantly, the complex relationship between mitochondria function and nuclear processes extends beyond the role of ROS.
MITOCHONDRIA HETEROGENEITY IN CANCER
In addition to their role as the bioenergetics center of the cell, mitochondria are central to a myriad of cellular functions including iron[37]and calcium homeostasis[38], metabolism of amino acids, lipids, nucleotides and carbohydrates, apoptosis, and a variety of signaling pathways[38-42]. Dysfunctions in many of these mitochondrial processes have been associated with cancer development[40,42]and chemoresistance[43].
Similar to nuclear genomic heterogeneity, metabolic heterogeneity is also widespread in tumors. The initialfindings by Otto Warburg of metabolic changes in cancer cells have been confirmed at multiple levels, fromin vitrocancer cell models toin situtumors in patients right before surgery[44,45]. Similar to the observations in genomic heterogeneity, these studies have revealed metabolic heterogeneity within different sections of the tumor[44]indicating that metabolic heterogeneity exists between tumors (inter-tumoral), within the tumor (intra-tumoral) and most likely also varies dynamically over time.
At the genetic level, comparisons using full genome sequencing in patient-derived pairs of cancer and normal tissues across multiple tumor types revealed the existence of somatic mtDNA mutations in a majority of tumors[46,47], with 31.1% of the tumors harboring multiple mtDNA mutations[47]. Unlike the nuclear genome,which contains two alleles of each gene, the mtDNA complement of a cell consists of hundreds to thousands of circular mtDNA molecules, allowing for different layers of mtDNA heterogeneity: alterations in mtDNA copy number, mutations in the mtDNA that occur in some but not all copies of the mtDNA genome within a cell (heteroplasmy), or mutations in the mtDNA that show dominance and accumulate until the mutant mtDNA becomes the only version present in the cell (homeoplasmy). Differences in mtDNA copy number,both increases and decreases of mtDNA relative to normal tissue, have been observed in many cancer types with some studies showing mtDNA copy number variation in up to 88% of tumors[48]. However, the role of mtDNA mutations or copy number variations as potential causative agents in cancer development have not been fully established due to the technological difficulties of manipulating the mtDNA genome. Studies in mice that have mtDNA from one strain and nuclear DNA from another strain (i.e., mice generated by mitochondrial-nuclear exchange) show effects in cancer progression models including changes in tumor size and metastatic burden[49], suggesting that the mtDNA can affect cancer progression.
INTERPLAY BETWEEN MITOCHONDRIA AND NUCLEAR FUNCTIONS
Genetic interconnections between the nucleus and the mitochondria are evident, since all but thirteen mitochondrial proteins are encoded by the nuclear genome. Associations between nuclear-encoded mitochondrial genes and tumorigenesis have been found, including mutations in several subunits of complex II, succinate dehydrogenase and isocitrate dehydrogenase, among other mitochondrial enzymes[50,51]. This nuclear control of the mitochondria by regulation of nuclear-encoded mitochondrial genes is termed anterograde signaling, and it is complemented by an equally important retrograde signaling system that allows the mitochondria to relay signals to the nucleus[52,53]. Retrograde signaling wasfirst identified via changes observed in transcription of nuclear genes in response to respiration defects[54]. Later studies established that the retrograde signaling response is a mitochondria quality control mechanism in which the cell senses different mitochondrial functions (e.g., ROS production, the TCA cycle, calcium levels, the unfolded protein response), and communicates the status of these functions to the nucleus via signaling cascades[52,53]. These retrograde signals activate diverse nuclear responses, setting in motion multiple pathways that regulate energy homeostasis, oxidative stress, and mitophagy, among other functions[52,53,55].
Importantly, mitochondria-dependent regulation of other nucleo-centric processes has started to emerge,including a role in regulation of the cell cycle.
THE MITOCHONDRIA MEETS THE CELL CYCLE
The eukaryotic cell cycle consists of four phases G1, S-phase, G2 and mitosis. These phases were historically defined by two genome-centric processes: DNA duplication (S-phase) and chromosome segregation (mitosis),interspersed with “gap” phases (G1 and G2) to allow for cell growth[56]. It is now understood that the cell cycle involves more than duplication and segregation of DNA. During a cell cycle cells must also grow and segregate their organelles and other cellular structures[57-59]. This duplication of the genome and increase in cell biomass, followed by the complex division of all cell contents to form two fully functional daughter cells requires a large amount of energy and metabolites. Links between metabolism and the cell cycle were identified early in the history of cell cycle research via genetic screens in budding yeast that identified Cell Division Cycle (CDC) mutants[60]. Several of the original CDC alleles, which cause cell cycle defects when grown at the non-permissive temperature, were later discovered to also result in reduced carbon metabolism and lower ATP production[61]. Conversely, mutations in cell cycle genes, such as the cyclin-dependent kinase CDC28 were found to also affect mitochondria biogenesis[62].
This metabolism-cell cycle connection has been studied in detail in budding yeast. Analysis of synchronously growing yeast populations uncovered cyclic changes of metabolism that associate closely with the phases of the cell cycle[63]. These studies determined that in budding yeast the metabolic cycle consists of three phases[64]: (1) oxidative respiration, marked by increased oxidative phosphorylation, increased ATP and amino acid production. This phase is aligned with entry and progression into G1 of the cell cycle; (2)reductive/building phase, characterized by an increase in glycolysis, increased production of nucleotides,nucleosides and ethanol. This phase occurs in synchrony with S-phase and mitosis; (3) reductive/charging characterized by production of complex carbohydrates for energy storage (e.g., glycogen, trehalose). This phase occurs during the end of mitosis and entry into quiescence (G0). The synchronicity of the cell cycle and metabolic cycle in budding yeast appears to be the result of a system of coupled oscillators, since the metabolic cycle can continue to oscillate in the absence of cell division[65,66]. Intriguingly, the expression of a number of cell cycle genes continues to oscillate with the metabolic cycle even in those cells that are not undergoing cell division, suggesting that the metabolic cycle can regulate cyclic expression of cell cycle genes independently of cell cycle controls[65].
Overall the evidence in budding yeast reveals an interaction between mitochondrial metabolism and the cell cycle. Evidence of a similar interaction in other organisms has only recently started to emerge.
MITOCHONDRIA DYNAMICS ARE REGULATED BY THE CELL CYCLE
Early studies on the connection between mitochondria processes and the cell cycle in human cells identified an increase in total mitochondria biomass that paralleled the increase in cell size during cell cycle progression[67]. Thefinding that mtDNA replication was not co-regulated with nuclear DNA replication[68], led to the idea that mitochondria and cell cycle processes were mostly unlinked. This view has changed recently as mounting evidence has shown that mitochondria biogenesis, morphology, dynamics and function are regulated by the cell cycle.
Mitochondria are highly dynamic organelles undergoing constantfission and fusion. These dynamics depend largely on several members of the dynamin family of proteins: mitofusin 1 and 2 (Mfn1/2) drive fusion of the outer mitochondria membrane and optic atrophy protein 1 (Opa1) mediates inner mitochondria membrane fusion, while dynamin-related protein 1 (Drp1) is required for mitochondriafission[69,70]. Mitochondriafission is also facilitated by four receptors that cooperate to recruit Drp1 to the outer mitochondria membrane:Mff, MiD49/51 and Fis1[71,72]. Importantly, mitochondria morphology and dynamics change in a cell cycledependent manner[73], with elongated mitochondria being dominant in G1[74]and short mitochondria being dominant in mitosis[75]. These changes in mitochondria dynamics during the cell cycle are controlled via regulation of mitochondria-dynamics proteins by the cell cycle machinery [Figure 1].
Mitochondriafission during mitosis in human cells is driven by Drp1, whose activity is increased in mitosis via phosphorylation by the mitotic cyclin Cyclin B1/Cdk1[76]. Drp1 phosphorylation in mitosis is promoted by another mitotic kinase, Aurora A, via phosphorylation of the small GTPase RALA and its binding partner RALBP1, which in turn bind to and facilitate Drp1 phosphorylation by Cyclin B1/Cdk1[77]. Mitochondriafission in mitosis is important for mitochondria segregation. Depletion of RALA or RALBP1 result in asymmetric segregation of the mitochondria to the two daughter cells, presence of mitochondria bridges during cytokinesis, and in some cases cytokinesis failure due to interference of the indivisible mitochondria mass with the cytokinetic ring[77]. In turn, Drp1 promotes mitotic exit (adaptation) of cells arrested in mitosis with the microtubule-stabilizing drug taxol via regulation of Cyclin B1 levels[78]. Similarly, ATP depletion by addition of 2-deoxy-glucose (2-DG) and sodium azide promotes mitotic exit in cells arrested in mitosis with the microtubule depolymerizing drug nocodazole, and this adaptation is also due to reduction in Cyclin B levels[79]. These results indicate a bi-directional crosstalk where the mitotic machinery increases Drp1 activity and mitochondria dynamics in mitosis, which in turn feedbacks to regulate mitosis[80]. Once the cells exit mitosis, Drp1 is targeted for degradation by APC/CCdh1[81], shifting the balance of mitochondria dynamics to favor mitochondria fusion.In addition to regulating mitochondria dynamics, cell cycle proteins also regulate respiration and other mitochondrial processes. Cyclin D1 represses mitochondria function by inhibiting nuclear respiratory factor 1(NRF1), a transcription factor that induces expression of a set of nuclear-encoded mitochondrial genes[82],and regulates gluconeogenesis[83]. A pool of Cyclin B1/Cdk1 localizes to the mitochondria, phosphorylates components of the OXPHOS machinery and increases their activity at the G2/M transition[84]. Some components of the spindle assembly checkpoint (e.g., Mad2, BubR1, p31-comet) have roles in insulin signaling[85], while others (e.g., Mps1, Survivin) localize to the mitochondria and regulate apoptosis[86,87].Together, these results indicate extensive regulation of mitochondria functions and/or cell metabolism by the cell cycle machinery.
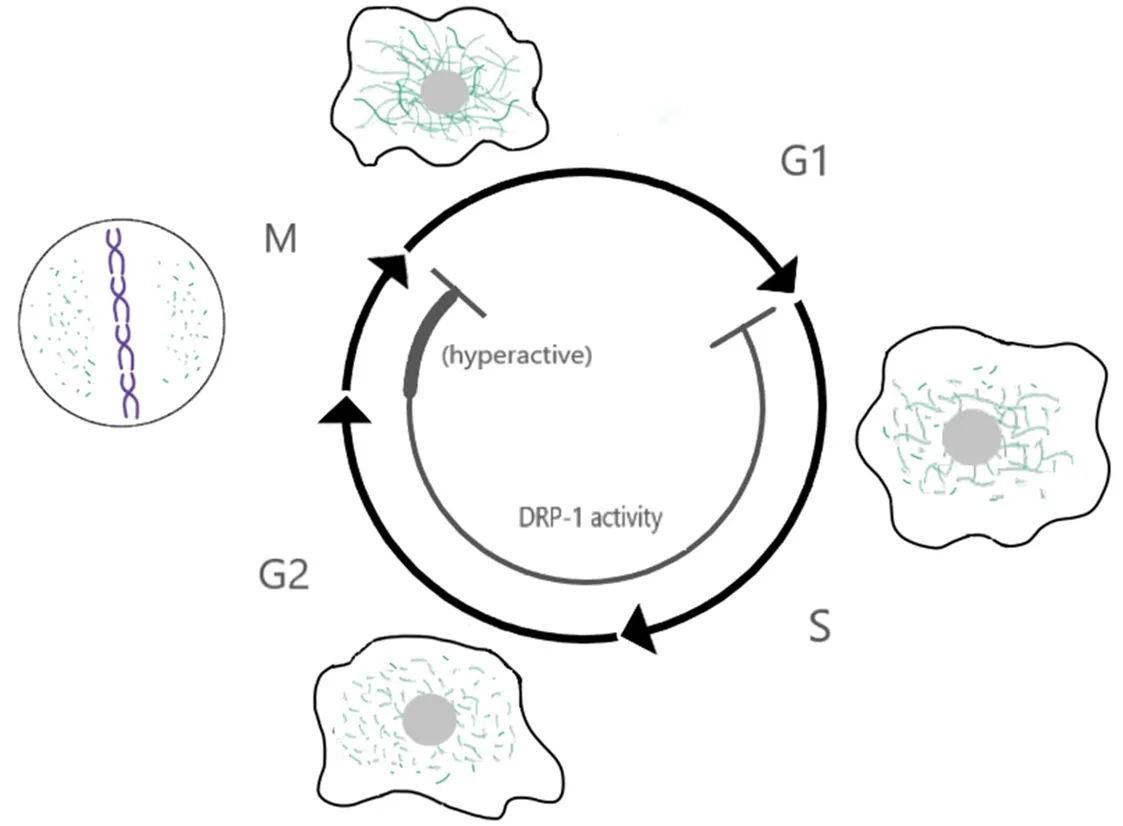
Figure 1. Crosstalk between mitochondria dynamics and the cell cycle. Model showing the changes in mitochondria dynamics and Drp1 activity throughout the cell cycle. Mitochondria fusion is favored in G1 and mitochondria fission is dominant in mitosis. This leads to the formation of a highly elongated and interconnected network during late G1, and small disconnected mitochondria in mitosis. Changes in mitochondria dynamics are regulated by the cell cycle machinery, for example mitochondria fission is favored in mitosis by Drp1 phosphorylation by the mitotic kinase Cyclin B1/Cdk1. Conversely, mitochondria fusion is favored in G1 due, at least in part, to degradation of Drp1 by the ubiquitin ligase APC/C-Cdh1 in early G1. In turn, these changes in mitochondria morphology regulate the cell cycle. Hyperfused mitochondria promote the G1/S transition, while inhibition of Drp1 induces a G2 arrest and failure to fragment mitochondria in mitosis can interfere with cytokinesis. These phenotypes have started to reveal a profound level of cross-talk between these two processes
THE CELL CYCLE IS IN TURN REGULATED BY MITOCHONDRIA FUNCTION
Increased mitochondria fusion after mitotic exit leads to the formation of a hyperfused mitochondria network in late G1 which promotes the transition from G1 into S-phase[74]. The molecular mechanism by which mitochondria hyperfusion promotes S-phase entry has not been completely elucidated. However,it appears that mitochondria hyperfusion and the accompanying increase in mitochondria respiration in late G1 promotes accumulation of the S-phase cyclin, Cyclin E[74]. Conversely, inhibition of respiration in G1 using the uncouplers FCCP or CCCP results in decreased Cyclin E accumulation and delay in S-phase entry[74,88]. This model is supported by an analysis of mitochondrial potential (ΔΨm) in a population of G1 cells which showed that G1 cells with low ΔΨm have a molecular profile corresponding to early G1 cells (e.g.,low Cyclin E, high p27Kip1), while G1 cells with high ΔΨm have a late G1 molecular signature (e.g., high Cyclin E, low p27Kip1)[89].
In addition to its role in promoting the G1/S transition, mitochondria dynamics also regulate the G2/M transition. Depletion of Drp1 results in a G2 arrest[90-92], due to the presence of DNA damage and activation of the DNA-damage kinases ATM and ATR[90]. A similar G2 delay accompanied by DNA damage is also observed after disruption of the Drp1 adaptor Fis1[93]. Disruption of other mitochondria functions, such as in aDrosophilaknockout of the mitochondria-specific form of RNaseZ[94]and in human cells depleted of mtDNA (rho0 cells)[95]also cause a G2 delay. Furthermore the G2 delay after Fis1 depletion correlates with low expression of the cell cycle transcription factor FoxM1 and its downstream mitotic genes, including Cyclin B1,suggesting that defects in mitochondria dynamics/function can lead to transcriptional inhibition of the G2/M transition[93]. The link between mitochondria dynamics and cell cycle gene expression is further strengthened by observations of a correlation between Drp1 expression levels and expression of cell cycle genes in different cancers, particularly genes expressed in G2/M[96]. Other metabolic alterations such as starvation and the subsequent induction of autophagy, or hypoxia have also been shown to regulate cell cycle progression[97].
Mitochondria dynamics/function have a role in the regulation of mitosis since Drp1 activity and ATP depletion promote mitotic exit in cells arrested in mitosis with microtubule-targeting drugs[78,79]. This exit from mitotic arrest when mitochondria function is compromised is due to premature degradation of Cyclin B1 by activation of the ubiquitin ligase APC/CCdh1[79]. These results indicate a complex cross-talk between mitochondria functions and the mitotic machinery, which has important implications for our understanding of the response of cancer cells to microtubule-targeting agents commonly used as cancer treatments (e.g., taxol,vinblastine). Additionally, other mitotic phenotypes are observed in cells with compromised mitochondria function, including amplification of centrosomes[90,95], abnormal centrosome positioning[98], chromosome misalignment[90]and multipolar spindles[95]. However, whether these phenotypes indicate a role for the mitochondria in the regulation of centrosome duplication or mitosis, or are merely consequences of the G2 delay and DNA damage observed in these cells has not been elucidated. Paradoxically, incubation with the Drp1 inhibitor Mdivi-1 seems to exert the opposite effect in cells damaged by x-rays. X-ray irradiation results in DNA damage, abnormal progression through mitosis (mitotic catastrophe), centrosome amplification and formation of micronuclei. In this scenario, incubation with the Drp1 inhibitor Mdivi-1 reduced the centrosome amplification and formation of micronuclei observed after irradiation[99].
Taken together, these results provide clear evidence of a bidirectional link between mitochondria dynamics/function and cell cycle progression at multiple phases. However, more research is needed to fully understand the extent of interaction between these processes and to understand the molecular underpinnings of this crosstalk.
OTHER LINKS BETWEEN THE MITOCHONDRIA AND NUCLEAR FUNCTION
As discussed previously, one of the best studied endogenous sources of genomic instability is the mutagenic potential of ROS, which can induce oxidative DNA damage[35,36]. Increased levels of ROS have also been shown to induce other types of damage such as telomere attrition and chromosome fusions[100]. However,other mechanisms by which mitochondrial dysfunction affects nuclear genome instability have started to emerge [Figure 2]. In budding yeast, loss of mtDNA leads to genomic instability and this was not correlated with defects in respiration, but rather with defects on the mitochondrial processing of iron-sulfur clusters[101].In addition, mtDNA can affect nuclear DNA through direct transfer of genes. This process, termed numtogenesis[102], was thought to be a rare event occurring at an evolutionary scale of millions of years.However, several reports have identified higher rates of numtogenesis in cancer cells. For example, a study identified mtDNA in the nuclei of up to 27.5% of cervical carcinoma cells compared to 0% of paired cells from the normal cervical epithelium[103]. Increased rates of numtogenesis were also observed via analysis of whole genome sequencing of adenocarcinoma samples[104]. Importantly, mtDNA integration into the nuclear genome can have important consequences such as activation of oncogenes[105].

Figure 2. Mitochondria functions impact nuclear functions. Model showing the different aspects of mitochondria function that have been shown to affect the nuclear genome at the level of gene expression (e.g., via calcium signaling pathways), genomic instability (e.g., DNA damage, telomere attrition) or epigenetic modifications (e.g., DNA methylation, histone modifications)
Another direct link between the mitochondria and genomic instability has been observed in cells that survive exposure to pro-apoptotic stimuli. Exposure of cells to a sub-lethal dose of the BH3-mimetic ABT737 results in partial mitochondria outer membrane permeabilization (MOMP), partial caspase activation and increased DNA damage[106]. One of the mitochondrial proteins, released from the mitochondria during MOMP is apoptosis inducing factor (AIF). AIF translocates to the nucleus and binds to the nuclear DNA, triggering condensation and fragmentation[107-109]. Importantly this nuclear translocation of AIF has also been observed in cells exposed to sublethal doses of oxidative stress[110], suggesting that release of mitochondrial proteins like AIF can result in DNA damage without triggering apoptosis.
In addition to introducing genomic instability, the mitochondria also modulates nuclear functions via retrograde signaling that regulates nuclear gene expression. Mitochondria play a role in the epigenetic regulation of the nuclear genome[111,112]. DNA methylation patterns have been shown to change in cells depleted of mtDNA (rho0)[113]and in cells with different mtDNA haplotypes[114]. Mitochondria also have a role in calcium regulation, and mitochondrial stress can induce calcium release, activating signaling cascades that can lead to different nuclear gene expression responses and phenotypic changes, such as increases in invasive behavior[115]. Similarly, reduction in mtDNA content in breast cancer cells activates a calcineurindependent pathway that induces phenotypic changes similar to the epithelial-to-mesenchymal transition(EMT) associated with higher cancer aggressiveness[116]. Retrograde signaling, alteration of epigenetic regulation, direct transfer of genetic material, and ROS-mediated effects demonstrate the myriad of ways that mitochondrial dysfunction can play a role in nuclear genome instability and function.
IMPLICATIONS OF MITOCHONDRIA-NUCLEAR INTERACTIONS FOR CANCER
Deregulation of cellular energetics is considered one of the emerging hallmarks of tumor development, while genomic instability has been established as an enabling characteristic of cancers[117]. The results discussed in this review provide evidence for a complex bidirectional cross-talk between mitochondria processes and nuclear processes involved in genomic maintenance, particularly regulation of the cell cycle. Identifying the molecular players involved in this cross-talk will not only open possibilities for the development of new cancer treatments, but it also reveals an unexpected complexity where genomic instability and defects in mitochondria function can synergize to accelerate cancer progression. That is, as cancer progresses and cell metabolism changes, these changes could lead to modifications in cell proliferation due to cell cycle dysregulation; while in turn modifications in the cell cycle or genomic instability can induce changes in mitochondria function, leading to a synergistic acceleration in the acquisition of cancer-associated traits.This synergism also accelerates the acquisition of heterogeneity whereby increases in genomic heterogeneity will promote heterogeneity of mitochondria function, and vice versa.
DECLARATIONS
Acknowledgments
The authors would like to thank Dr. Sid Das and the Das lab for continuous support and helpful discussions.
Authors’ contributions
All authors contributed to the writing of this review. In addition Herrera EL prepared thefigures.
Availability of data and materials
Not applicable.
Financial support and sponsorship
Work in our laboratory is partially funded by UTEP’s Research Incentive Program and a grant to LADM from BD-Biosciences. Herrera EL is an NIGS RISE-scholar funded by grant R25GM069621-11. Azzam SZ is an NSF S-STEM scholar funded by grant DUE1565063.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2018.
 Journal of Cancer Metastasis and Treatment2018年8期
Journal of Cancer Metastasis and Treatment2018年8期
- Journal of Cancer Metastasis and Treatment的其它文章
- AUTHOR INSTRUCTIONS
- Unmasking tumor heterogeneity and clonal evolution by single-cell analysis
- Centrosome aberrations and chromosome instability contribute to tumorigenesis and intratumor heterogeneity
- Increased ARF6 activation correlates with HGF stimulation in non-invasive prostate cancer cells
- Expression and reguIation of aIdehyde dehydrogenases in prostate cancer
- Extracellular control of chromosomal instability and maintenance of intra-tumoral heterogeneity