
Expression and reguIation of aIdehyde dehydrogenases in prostate cancer
2018-11-21 01:58:18AliIbrahimMariaSadiqFionaFrameNormanMaitlandKlausPors
Ali I. M. Ibrahim, Maria Sadiq, Fiona M. Frame, Norman J. Maitland, Klaus Pors
1Institute of Cancer Therapeutics, School of Pharmacy and Medical Sciences, Faculty of Life Sciences, University of Bradford, West Yorkshire BD7 1DP, UK.
2Department of Pharmacy, Al-Zaytoonah University of Jordan, Amman 11733, Jordan.
3Cancer Research Unit, Department of Biology, University of York, Heslington, North Yorkshire YO10 5DD, UK.
Key words:Aldehyde dehydrogenase,retinoic acid,prostate cancer,castration-resistant prostate cancer,cancer stem cells,multidrug resistance
ABSTRACT The functional role of aldehyde dehydrogenases (ALDHs) in prostate cancer remains an area of some controversy. Many studies have used high ALDH functional activity to isolate putative cancer stem cells with tumour-initiating and propagating properties, while evidence is also emerging about the involvement of specific isoforms in migration, invasiveness and metastasis. Identification of specific ALDH isoforms, which contribute to both drug resistance and aggressiveness of the disease remains a challenge within the complex heterogeneity of prostate cancer. The purpose of this perspective is to dissect functional roles for ALDH in the tumour microenvironment and to evaluate the potential of the ALDH gene family as biomarkers and/or targets for therapeutic intervention.
INTRODUCTION
Prostate cancer (PCa) is the most common cancer affecting men in the developed world. In the UK alone,over 47,000 new cases are diagnosed and more than 11,000 cancer related deaths are registered every year (Prostate Cancer UK, 2016). PCa is often present in the absence of apparent symptoms for many years, and so is considered to be slow-growing,however this is not true for all PCa’s. Whilst the underlying cause of PCa is not fully understood, the initial stages of PCa frequently depend on androgens for cellular proliferation. If radiotherapy or radical prostatectomy cannot be used to eradicate or remove the tumour, then it is effectively treated by androgen deprivation therapy (ADT)[1,2], especially if the tumour has escaped the capsule. However, the tumour invariably relapses in most patients after ADT, leading to an aggressive form of PCa known as castration-resistant prostate cancer (CRPC), which remains an untreatable disease[1,3].

TabIe 1: Tissue distribution, subceIIuIar distribution and substrates of human aIdehyde dehydrogenases
The tumour microenvironment (TME) exerts a strong hold on tumour initiation, progression and metastasis[4]. TME is a general term encompassing a complex heterogeneous environment which includes inflammatory cells, blood vessels, extracellular matrix[5]andfibroblasts (stroma). In normal prostate homeostasis, a controlled interaction between nonepithelial components such as stroma and epithelial cells contributes to normal epithelial proliferation,differentiation and migration[5,6]. When prostate epithelial cells have acquired a malignant phenotype,this crosstalk between prostate epithelium and stromal cells is perturbed[6]. As a consequence,stromal cells play a critical role in activating cellular events within the TME that sustain and support cancer proliferation and metastasis[4,7]. Multiple studies of cell signalling associated with androgen[8],Hedgehog[9], fibroblast growth factor (FGF)[10], Src family kinase[11], transforming growth factor-β (TGF-β)[12],Integrin[13]and Notch[14]pathways, implicate the TME,however many such observations are derived using a mixture of both human and mouse models in which the TMEs are radically different. Accordingly more careful attention is required to evaluate the impact of the TME.
Within a tumour, the entire population of replicating cancer cells has been hypothesised to be derived from a small subpopulation of cancer stem cells(CSCs) or tumour initiating cells (TICs)[15]. CSCs have the ability to both self-renew and to produce progenitor and differentiated cells, generating phenotypically diverse tumour cell populations[16].The stem cell microenvironment (SCME) is a specific anatomic location (or “niche”) where stem cells (SCs)are located, and the interplay between SCs and these niches can regulate the dynamic process of SCs’role in tissue generation, maintenance and repair[17].Several factors affect SC regulation within the SCME,including the interactions of SC with each other,with differentiated cells, and with extracellular matrix components[18]. Dysfunction of a cellular process or signalling pathway within the SC niche could contribute to the evolution of a CSC[19]. Although the presence of this CSC niche could pose obstacles for the treatment of PCa, it has also been proposed that the CSC niche also provides a potential target for biomarker and drug discovery[20-22].
Aldehyde dehydrogenases (ALDH) have been exploited as selective markers for CSCs and have been assigned potential functional roles in differentiation, self-protection and expansion[23]. The ALDH superfamily consists of 19 genes with distinct chromosomal locations, which are found across 11 families and 4 subfamilies[23-25]. These enzymes have a varied tissue and organ distribution[26-28]and are localised in the cytoplasm, mitochondria, nucleus,and endoplasmic reticulum[23,24]. ALDH isoforms show distinct substrate specificity[26,29,30], and are NAD(P)+ dependent [Table 1]. Their major role is the detoxification of endogenous and exogenous molecules, via oxidation of aldehyde substrates to their corresponding acids. This catalytic oxidation is a fine-tuned reaction evolved to protect cells from the harmful effects of highly reactive aldehydes and maintains cellular homeostasis[24,25,31]. Vital functions include protection of cells from oxidative stress (e.g.,reactive oxygen species, ROS) and promotion of retinoic acid (RA) metabolism and signalling[32].
Mutations and polymorphisms in ALDH genes lead to a loss-of-function that are associated with various human pathologies[33-39], which supports their important biological function. Plentiful studies have described the expression of ALDHs in human tissues,however their expression profile and functional activity is poorly understood within the TME. As a consequence of high and abundant expression,ALDHs have been considered to be biomarkers of specific tumour types[40-45]. Human ALDHs are among the regulatory proteins that catalyse the retinoic acid(RA) pathway, which has been linked with “stemness”characteristics[45]. The ALDH1A subfamily members have also been identified in a wide-range of human CSCs, and their expression has been associated with poor prognosis in patients with several cancer types including PCa[46-54].
ALDH EXPRESSION AND REGULATION IN PROSTATE CANCER
The rate and frequency of PCa progression varies considerably between individuals, ranging from relatively slow (indolent, non-invasive) in some patients whilst in other cases the disease is more aggressive and results in rapid metastasis[55]. At present, PCa is diagnosed at first by monitoring levels of serum prostate-specific antigen (PSA) and digital rectal examination[55]. However there is a substantial overlap in PSA levels between patients with benign prostatic hyperplasia (BPH) and patients with PCa[55]. About 25% of cases with PCa display no increase in serum PSA levels and thus must be detected by other methods[55], such as diagnostic needle biopsies and MRI scans. Furthermore, it is crucial to determine indolent from aggressive forms of PCa, to offer patients earlier diagnosis and better treatment options. This is neither currently possible nor routine. In this regard, more detailed, in-depth understanding of the correlation between ALDHs and PCa progression may provide alternative biomarkers for disease diagnosis and treatment.
As indicated above, a complex interplay of PCa with the surrounding stroma, androgen receptor(AR) signalling, epithelial-to-mesenchymal transition(EMT) and other signalling pathways within the TME support progression of the disease. Stromal cells such asfibroblasts and myofibroblasts are involved in hormone signalling, contributing to stromal-epithelial interactions in the primary tumour setting[56-58].For example both stromal and epithelial ALDH1 expression, measured using IHC, have been shown to be a potential biomarker for breast cancer[59]. The epithelial and stromal ALDH1 expression (detected in 43% and 69% of benign breast biopsies, respectively)was associated with a predicted increase in the risk of breast cancer. However, as with many earlier studies[45]on profiling ALDHs in clinical specimens,no information is available to ascertain which ALDH was overexpressed from the subfamily (ALDH1A1,1A2, 1A3, 1B1, 1L1, 1L2).
In PCa, several ALDH isoforms (1A1, 1A3, 3A1, 3A2,4A1, 7A1, 9A1 and 18A1) have been found to be overexpressed[15,60-68], but only a few isoforms appear to play critical roles in PCa. In a recent proteomic study[69], ALDH1A3 expression was in part controlled via miR-187, as downregulation of this microRNA led to induction of ALDH1A3, while re-introduction decreased ALDH1A3 expression in PC-3, DU145 and LNCAP prostate cancer cells. Some ALDHs may also contribute to regulation of AR pathways,with implications for normal prostate development,prostate carcinogenesis and progression to androgen-independent disease[70-73]. AR is expressed in almost all primary PCas[74-76]and the transition from a localised hormone-naïve to a castrationresistant phenotype is based on a complex interplay of signalling molecules attributed to aberrant AR signalling[73,77-79]. Raised PSA suggests that AR function is still active but abnormal in CRPC[80], due to a number of different mechanisms including AR amplification[81], AR gain-of-function mutations[82],intracrine androgen production[83], elevated levels of AR cofactor that sensitises cancer cells to low levels of androgens[84], ligand-independent activation of AR by growth factors and cytokines[85]and constitutively active messenger ribonucleic acid (mRNA) spliced variants of AR[86]. Consequently, AR remains a critical factor in the progression of early-stage PCa to CRPC.
ALDH1A3 is androgen responsive in human epithelial LNCaP cells since its expression was 4-fold higher after treatment with dihydrotestosterone (DHT),which indirectly affects both AR regulation and cell differentiation[59]. ALDH1A3 has also been correlated with AR signalling pathway in primary PCa tissue where expression was consistent with luminal layer localisation[65]. Significantly, the study also showed that knockdown of ALDH1A3 led to substantial reductions in proliferation rate and the invasive ability of PC-3 cells. However, the regulation of ALDH1A3 expression is likely to be multifactorial[87].
Outside the ALDH1 family, strong association of ALDH3A1 with PCa progression has also been demonstrated in both immortalised cancer cell lines and tumour xenografts[61]. In clinical tissues ALDH3A1 was detected in intra-epithelial neoplasia,with elevated levels in carcinomas in the absence of expression in normal prostate glands. Finally,in comparison with the paired local carcinomas,ALDH3A1 was upregulated in both lymph node metastatic tumours and was detectable in bone metastatic PCa.
ALDH7A1, which has also been related to the stemness of CSCs[88], is mainly localised in the cytosol, but it has also been found expressed to a lesser degree in the mitochondria and nucleus[32,45].In addition to catalysing aldehyde metabolism,ALDH7A1 also plays a role in protecting tissues from the damaging effects of osmotic stress[89]while mutation of the ALDH7A1 gene has been related to pyridoxinedependent epilepsy[90,91]. In cancer, ALDH7A1 is expressed in nodular melanoma (NM)[92], ovarian[93]and lung cancers[94]while in PCa the isoform has been shown to be involved in intra-bone growth and induced bone metastasis[64]as well as zoledronic acid resistance[95]. Gene expression profiling supports the involvement of ALDH7A1 in multiple molecular pathways related to the metastatic process in PCa[96].
EVIDENCE FOR EPIGENETIC CONTROL OF ALDHS
PCa can be initiated by genetic or epigenetic alterations, including DNA methylation in the promoter region of genes, normally linked to transcriptional silencing[55]. Epigenetic changes including DNA methylation and histone modifications of tumour suppressor genes (TSGs) preferentially occur in the early stages of cancer progression[55].The promoter region of ALDH1A2 in primary PCa specimens has been shown to be densely hypermethylated in comparison to normal prostate tissues[97]. This observation is supported by another study that showed a low/absent expression of ALDH1A2 in PCa in formalin-fixed paraffin embedded sections compared to elevated levels of expression in normal prostate tissue[98]. On this basis it was suggested that ALDH1A2 act as a TSG in PCa,and that its epigenetic regulation could differentiate normal prostate cells from malignancy. In contrast,ALDH1A3 has been demonstrated to be an androgen responsive gene[67]whose induction contributes to the conversion of retinol to RA with potential for supporting cellular proliferation[55]. Hypermethylation of the ALDH1A3 promoter region in clinical tissues has also been detected[99], but this study used a relatively small sample size (n= 24) and did not distinguish between methylation of basal and luminal PCa cells. Although larger studies are required, it is possible that methylation of the promoter regions of ALDH1A2 and ALDH1A3 could be used as a marker for PCa detection[55].
ALDH EXPRESSION IN CSC MICROENVIRONMENT
Growing evidence strongly supports initiation of PCa from CSCs residing within a basal niche[100-105]. In xenotransplantation experiments, less than 100 TICs are needed to generate a new tumour in mice and these cells exhibit a basal phenotype[106]. Furthermore,using human tissue biopsies the prostate SC markers CD44+, α2β1-integrinhigh and CD133+ have been used to identify and isolate prostate CSCs with selfrenewal capacityin vitro[100]. Additionally, there are other important markers that have been used to identify and isolate PCa SCs. ATP binding cassette(ABC) transporters which are proteins that play a vital role in the ef flux of drugs have also been used to enrich CSCs. However, CD44+, α2β1-integrin high and CD133+ ABC transporters are also expressed in normal SCs[107,108]which emphasises the need to employ at least two markers to avoid cross reacting populations of cells[107]. A growing body of evidence suggests that the functional activity of ALDHs can be used to identify and purify CSCs from e.g. breast[109],ovary[110], lung[111], colon[112], pancreas[113]and prostate cancer[114]. At present it is unclear if ALDH expression is significantly different between normal SCs and CSCs, hence more research is required to understand if any isoforms could be more predictive than e.g. CD44+, α2β1-integrin high and CD133+used as a PCa SC gene-expression signature[115].
ALDHs expressed in SCs are members of the ALDH1 family (1A1, 1A2, 1A3, 1L1, 1L2), ALDH2, ALDH3A1,ALDH4A1 and ALDH7A1, which have all been linked with various critical roles including chemo-protection,DNA damage and regulation of the cell cycle[24].The Aldefluor assay has frequently been used to identify and isolate CSCs, but as this assay does not distinguish between different isoforms many studies suffer from a lack of knowledge of the contributing ALDHs to the stemness of the isolated subpopulations with tumourigenic properties. However, some studies have shown that e.g. the ALDH1A1 isoform positively correlates with the expression of CSC surface markers CD133[116]and CD34[117]with utility in characterising liver CSCs and leukaemia SCs,respectively. The association of ALDH3A1 has also been reported in PCa progression[61]. Stem cell-like cells from DU145 cells have elevated expression of ALDH3A1 compared to non-stem counterparts, and the stem cell-like population generated xenograft tumours with aggressive features[118].relaxed by the action of histone acetyltransferase(HAT) or methyltransferase activity[143], facilitating the recruitment of transcriptional machinery which stimulates RA responsive gene transcription[144,145].
ALDHS AND THE RETINOID SIGNALLING PATHWAY
Retinoic acid (RA, all-trans retinoic acid (ATRA),tretinoin) the physiologically active metabolite of vitamin A (retinol) is a potent regulator of signalling pathways during embryonic development[119]. RA is necessary for adult tissue homeostasis and acts through nuclear retinoic acid receptors (RARs)[120],with diverse immune modulatory roles[121,122], role in spermatogonial differentiation[123], and cancer[124-126].RA is endogenously produced from retinol (vitamin A)in two subsequent metabolic steps [Figure 1]: thefirst step is the retinol oxidation to retinaldehyde, which is catalysed by several alcohol dehydrogenases (also known as retinol dehydrogenases)[127,128]. The second step is the oxidation of retinaldehyde to retinoic acid,which is an irreversible step carried out by ALDHs(also known as retinal dehydrogenases)[129]. At least four ALDH isoforms, ALDH1A1, 1A2, 1A3 and 8A1,have been shown to be responsible for the oxidative formation of retinol to RA[128,130-132]. ALDH1A3 appears to be the most catalytically efficient enzyme for RA oxidation and has no apparent capacity to metabolise cis-retinal substrates[133]. The involvement of ALDHs in RA synthesis underpins their vital function in pathways associated with cell proliferation,differentiation and survival[87].
The RA biosynthetic pathway is likely to be suppressed or activated depending on the local prostate microenvironment[146-148]. The effect of RA has been investigated in normal and malignant prostate tissues[129,149]. Differential expression of RA was demonstrated in normal prostate, BPH, and prostate carcinoma tissues[129]. For example it was found that endogenous retinol levels were 2-fold elevated in BPH compared to normal and PCa tissue while RA levels were found 5-8 times lower in PCa tissue compared with the other two tissues. The authors speculated that the reason for this elevated level of retinol in BPH could reflect (1) a reduced activity of the dehydrogenase that metabolises retinol to retinal or (2) uptake from serum that metabolises retinol to retinal. A possible cause for the reduced level of RA in PCa could be a more rapid degradation of RA by cytochrome P450 enzymes[150].
In addition, RA also has variable effects on PCa signalling pathways, either directly or indirectly by regulating certain transcriptional factors such as NR2F1[151]and RA receptor responder 1 (RARR1)[152]since RA represses invasion and SC phenotype by induction of metastasis suppressors RARR1 and latexin (LXN) in PCa[153].
The synthesised RA binds to nuclear RAR and retinoid X receptor (RXR) forming a heterodimeric complex, which binds to RA response elements(RAREs), leading to downstream regulation of gene expression and cell differentiation events[134-137]. RA and 9-cis-RA (isotretinoin) bind to RARs, whereas only 9-cis-RA can bind to RXRs[23]. In response to RA synthesis, cellular retinoic acid binding protein(CRABP) shuttles RA to the nucleus where it binds to the RAR/RXR heterodimer[138,139]. This subsequently results in the dissociation of co-repressors NCoR,SMRT and HDAC complex[140]and allows coactivators such as SRC/p160 family, p300/CBP and CARM-1 to bind[141,142]. The chromatin structure is
Retinoids are used as cancer treatment, in part due to their ability to induce differentiation and arrest proliferation. In the clinic, RA has been clinically investigated in PCa as single treatment[154], or with other agents in attempts to produce synergistic effects[155-157]. However, delivery of retinoids presents a challenge because of the rapid metabolism and the epigenetic alterations that can render cells retinoid resistant[158]. This poses new challenges rather than solutions. ALDH1A3 expression is regulated by many factors and is linked to many metabolic pathways including glycolysis and retinoid signalling, which has been recently reviewed[87]and hence not further discussed here.

Figure 1: Aldehyde dehydrogenases (ALDHs) expression and function in the tumour microenvironment. ALDH expression in cancer stem cells (CSCs) and differentiated cells have been linked with several cellular processes including glycolysis/glucogenesis and amino acid metabolism, which are likely to be affected by the local microenvironment including impact by hypoxia (A, B). Various ALDH isoforms have been shown to be regulated by e.g. tumour suppressor genes, oncogenes and microRNAs, however a well-documented functional role is linked with the retinoic acid (RA) pathway resulting in transcriptional activation of a number of genes important in cell differentiation (C).High ALDH expression is frequently used as an endogenous marker that in combination with cell surface markers can be used to isolate CSCs (D). More research is required to understand how ALDH activity may contribute to signaling pathways, maintenance of CSCs and contribute to tumour aggresiveness (D, E)
The relationship between AR and ALDH1A3 has been studied in both normal and tumour tissues, to understand the exact mechanism of their interaction,and its relationship to the role of ALDH1A3 as a marker of CSCs in several tumour types. In breast cancer, a potential link between ALDH1A3 expression and RA signalling contributed to an increase in the rate of cancer progression[159]. In human epidermal keratinocytes, it has been shown that regulation of RA metabolism involved the transcriptional activation of only ALDH1A3 amongst a panel of ALDH genes[160].ALDH1A3 activity induced by RA-regulated genes has been proposed to play a role in establishing a unique transcriptional profile that favours the CSC phenotype[161,162]. Conversely, a recent study revealed that ALDH1A1, 1A2 and 1A3, were downregulated in the undifferentiated embryonal cancer Wilms’ tumour 1 (WT1) resulting in inhibition of RA synthesis[163].Blumet al.[164]investigated the regulation of both RA and ALDH1A3 in the urogenital sinus epithelium(UGE), which contains primitive foetal prostate cells.A number of the major regulators of the RA receptor,including ALDH1A3 were up-regulated in both primitive populations of adult and foetal prostate SCs,with 10-fold increased ALDH activity in adult prostate SCs compared to cell population (Sca-1Neg) with no regenerative potential. In addition, expression of CRABP, which transports RA into the nucleus to bind RA receptors was 47-fold up-regulated in the UGE,as confirmed by qPCR analysis, and may indicate the potential of these cells to differentiate. In the context of PCa, ALDH1A3 might play a significant role in the CSC niche of the TME, thereby contributing to a survival mechanism.
THE CSC NICHE, SIGNALLING PATHWAYS AND POTENTIAL FOR THERAPEUTIC INTERVENTION
Cancer cells acquire a more invasive and migratory phenotype through EMT[165-168]. Cell adhesion is reduced in early metastatic PCa by downregulation of expression of E-cadherin and β-catenin(characteristically expressed in normal epithelial cells)[169]. In contrast, the expression of N-cadherin(characteristically expressed in mesenchymal cells)is upregulated[170]. In clinical specimens there is lower E-cadherin and β-catenin expression and higher N-cadherin expression in higher grade PCa compared to lower grade PCa[171-174]. However restoration of elevated E-cadherin expression and β-catenin was seen in metastatic cells deposited in the bone[175], implicating expression control rather than total E-cadherin gene loss.
The Wnt/β-catenin signalling pathway plays a significant role in maintaining the stemness of PCa[176,177]. In radioresistant ALDH+ (identified by Alde fluor assay) prostate progenitor cells, activation of EMT and the Wnt/β-catenin signalling pathways has been demonstrated. In this study, ALDH1A1 gene expression was regulated by the Wnt signalling pathway and correlated with simultaneous expression of β-catenin in whole prostate tumour specimens[178].Encouragingly, inhibition of the Wnt pathway (by siRNA knockdown or the tankyrase inhibitor XAV939)resulted in reduced ALDH+ tumour progenitor population and radio-sensitisation of cancer cells[178].The link between ALDH1A1 and β-catenin has also been demonstrated using spheroidal aggregates in a xenograft model comprised of ovarian cancer cells with stem cell characteristics[179]. In this study, β-catenin knockdown decreased ALDH1A1 expression, which subsequently led to inhibition of tumour growth and metastasis.
As described above, ALDH7A1 is highly expressed in primary PCa tissue[15,88]. ALDH7A1 knockdown decreased the stem/progenitor cell subpopulation in the human PCa cells and tumour migration abilityin vitro[88]. The activity was correlated with increased TGF-β signalling, which strongly induced ALDH7A1 activity while the activity could be inhibited with a TGF-β signalling antagonist[88]. Overexpression of the TGF-β signalling pathway correlates with poor clinical outcomes in PCa. TGF-β promotes tumour progression by stimulating the metastasis and angiogenesis[180].
As with many other studies, investigation of ALDH+cells isolated from both PCa cell lines and primary cells have shown self-renewal, colony forming capacity and tumourigenicity. ALDH expression correlated with CD44 and α2β1-integrin expression as well as phosphorylation of the transcription factor STAT3.Galiellalactone, a potent and specific inhibitor of STAT3 signalling, reduced ALDH1A1 expression and subpopulation of ALDH+ cells following treatment of DU145 PCa xenografts. This study highlighted the role of the STAT3 signalling pathway in putative prostate CSCs and further supports STAT3 as a potential therapeutic target[181]. In a separate study using primary tumour cells, STAT3 inhibition resulted in both cell death and CSC differentiation, resulting in a loss of both colony forming and tumour initiating capacity[182].
ALDH ASSOCIATED DRUG RESISTANCE IN THE TME
A number of studies have linked ALDH expression with chemotherapy resistance, although the underlying mechanisms are not well understood.Whilst chemotherapy reduces the bulk of a tumour,it also enriches the previously described CSC population[183-185]which are not susceptible to anti-mitotic drugs currently approved for clinical use. Although evidence is not available in PCa,CSCs have been shown to be highly resistant to both radiotherapy and chemotherapies including temozolomide, gemcitabine, etoposide, carboplatin,paclitaxel, fluorouracil, mitoxantrone, daunorubicin and cyclophosphamide (CPA)[186-200], contributing to tumour recurrence and metastasis. There are several possible mechanisms for CSC resistance to cancer therapy. Firstly, CSCs are slow-proliferating cells in a quiescent state and thus resist drugs primarily designed to target rapidly dividing cells[201]. Secondly,CSCs resist irradiation because of increased activation of the DNA damage checkpoint response,as exemplified in a recent study of glioblastoma CSCs[202]. Thirdly, increased expression of ABC transporters protects CSCs from high concentrations of drugs[203], as demonstrated by removal of Hoechst stain in analysis of side populations[204,205]. Lastly, high ALDH expression is likely linked to metabolic and detoxifying mechanisms, supporting a role as chemoprotecting enzymes[201].
Early studies first demonstrated a chemo-resistant role for ALDHs in a CPA resistant L1210 leukaemia cell line[206]. This study showed that high levels of ALDH activity were found in L1210 cells and that treatment with disulfiram (ALDH inhibitor) reversed the resistance phenotype of the cells to CPA. A subsequent study confirmed the role of ALDH-mediated CPA resistance in medulloblastoma[207].Similar studies demonstrated that high ALDH activity indicates CPA resistance in cancer and CSCs[208].Accordingly, inhibition of ALDH activity can in principle serve to sensitise CSCs to drugs such as CPA[209]. More specifically, ALDH1A1 and ALDH3A1 were both shown to inactivate CPA analogues[210,211].
The sphere forming cells (a common property of CSCs), from the sarcoma cell line MG63 were significantly insensitive to doxorubicin and cisplatin treatment compared with monolayer adherent counterparts. The sarcosphere cells with high ALDH1 activity were proposed as candidate sarcoma SCs, in which efficient drug detoxification is likely to have contributed to generation of a chemo-resistant CSC phenotype[191]. Furthermore, high ALDH expression in CSCs has shown chemo-resistance in both breast CSCs[190,212]and head and neck squamous cell carcinoma (HNSCC) SCs[213], where ALDH expression was associated with high Snail expression, a marker of EMT. Knockdown of Snail expression significantly decreased the expression of ALDH1 whilst blocking the tumorigenic abilities of CD44+ CD24- ALDH1+ cells[213]. Although many chemotherapeutic drugs are less effective in ALDH-expressing cancer cells, the underlying mechanisms are poorly understood. None of the drugs contain aldehyde functional groups that are direct substrates for biochemical reactions with ALDHs, but esterase activity has been shown for some of these enzymes,which potentially provides an ALDH mediated resistance mechanism for drugs such as the taxanes. Phase 1 metabolism resulting in short lived aldehydes as illustrated for CPA are direct substrates for ALDH detoxification, providing a potential resistance mechanism in ALDH+ expression cells including CSC population within the TME [Figure 2].Drug resistance can be reversed by co-treatment with an ALDH inhibitor such as DEAB. For example,doxorubicin, paclitaxel and radiotherapy resistance in breast cancer cell lines has been reversed following treatment with DEAB or RA[190].
ALDH, HYPOXIA AND TME
Hypoxia is not only a major feature of the tumour microenvironment but is also a potential contributor to the multidrug resistance (MDR) and enhanced tumourigenicity of CSCs[214]. Within the proposed hypoxic CSC niche, the cells are surrounded by an acidic microenvironment that activates a subset of metastasis promoting proteases such as MMPs and cathepsins[215]. As a consequence of poor angiogenesis and the inaccessible location,hypoxic cells are exposed to insufficient drug concentrations, which promote the survival of a drugresistant sub-population of cells. The lower oxygen tension increases resistance to radiotherapy and as discussed above, also enriches CSC niche within the TME. Hypoxia-activated prodrugs (HAPs) have been investigated for several decades and have shown considerable promise in combination with chemotherapy or radiotherapy, but no HAPs have yet been approved for clinical use. Unravelling the PCa microenvironment is likely to offer new insight and opportunities to molecularly stratify patients for treatment, based on their tumours’ hypoxic signature,including analysis of enzymes with oxidase and/or reductase functionality. Prostate tumours are considerably hypoxic as discussed in this thematic issue[216]and enzymes such as ALDHs are likely to be expressed differentially within the TME due to different pressures including hypoxic stress and types of cells such as MDR and CSCs.
The limited sensitivity of hypoxic tumours to radiotherapy may in part be related to CSCs residing in the hypoxic niche. Primary human PCa samples express both elevated levels of ALDH1A1+ and hypoxia inducible factor 1 alpha (HIF-1α), which have been linked to radioresistance[217,218]. A recent study[219]demonstrated that irradiation enriched the CSC population of DU145 and PC-3 cells. The irradiated cells were shown to possess elevated ALDH functional activity as well as DNA damage response activity, andin vivothe irradiated ALDH+cells were shown to maintain their tumorigenic properties, suggesting these might be radioresistantin vivo. Furthermore, in primary human prostate tumours, IHC analysis revealed co-localisation of ALDH1A1 and HIF-1α expression, implying that a subset of ALDH+ cells resides in the hypoxic niche and emphasising the need to target these to effectively eradicate heterogeneous prostate tumours.In other tumours, for example radiation resistant mesenchymal glioma, the SCs (MGSCs) possess elevated glycolytic activity and ALDH activity, in contrast to benign proneural SCs. Expression of ALDH1A3 was increased in clinical high-grade glioma compared with low-grade glioma or normal brain tissue[220]. Encouragingly, although the MGSCs were very aggressivein vitroandin vivo, the pan-ALDH inhibitor DEAB significantly reduced cellular proliferationin vitro. This investigation suggested that two subtypes of MGSCs, harbouring distinct metabolic signaling pathways, constitute intratumoural glioma heterogeneity. ALDH1A3 was proposed to play an important role in the glycolysis pathway, via catalytic metabolism of acetaldehyde to acetate that is in turn linked to the tricarboxylic acid (TCA) cycle[220]. The glycolysis pathway is interesting because of the link to the TME and what is defined as the “Warburg effect”.A recent study[221]reported on the mitochondrial pyruvate carrier 1 (MPC1) gene in knockout studies using CRISPR/Cas9 technology in RM-1 murine PCa cells. The MPC1 gene knockout cells revealed a metabolism reprogramming to aerobic glycolysis with reduced ATP production, increase in cell migration and resistance to both chemo- and radiotherapy.In addition, the MPC1 knockout cells expressed significantly higher levels of the stemness markers Nanog, HIF-1α, Notch1, CD44 and ALDH.

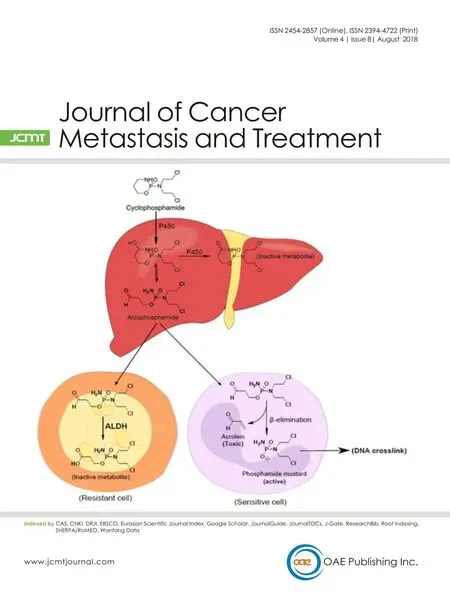
Figure 2: Cytochrome P450 (CYP) activation of cyclophosphamide (CPA). Initial hydroxylation of CPA in the liver by CYP isoforms leads to generation of aldophosphamide, an intermediate which is a substrate for aldehyde dehydrogenases (ALDHs) metabolism. If aldophosphamide enters circulation it is very likely to be detoxified in ALDH-expressing cells including cancer stem cells (CSCs), but not in cancer cells with low or absent ALDH expression
The latter study provides an alternative route for therapeutic intervention, focussed on reprogramming glycolytic pathways. ALDHs such as the 1A3 isoform could be a key player in such therapeutic intervention.However, as we[45]and others[46,87,222]have discussed previously, the expression of ALDHs in normal tissue expression remain a stumbling block towards a credible clinical therapy. However, advances in drug delivery technologies could in the future enable administration of an ALDH inhibitor, which is potently selective for a specific isoform. For example, a recent report[223]indicate that the latter might be achieved in combination with radiotherapy, or as an option to sensitise heterogeneous prostate tumour responses to docetaxel.
CONCLUDING REMARKS
The number of papers that report ALDH expression in the context of cancer is largely attributable to the use of the Alde fluor assay as a means to identify and isolate subpopulations with particularly stemness characteristics. However, selected ALDH isoforms are also emerging as critical players in chemoand radioresistance and a signature of tumour aggressiveness in conjunction with cells capable of migration, invasion and metastasis. Still, as is clear from this review of ALDH expression and function in PCa and other recent reviews[45,46,87,222],the ever increasing number of publications that reveal inconsistent and sometimes contradictory information is not helpful in clarifying ALDHs as potential biomarkers of specific cancer types or CSC population; e.g., many early studies that reported on ALDHs, utilised antibodies that only stained for e.g.ALDH1 but were not selective for 1A1, 1A2, 1A3,1B1, 1L1 or 1L2. Equally the Aldefluor assay is not isoform-selective and has contributed to inefficient validation of these enzymes. Furthermore, previous studies were carried out when the understanding of cancer cell subtypes, and the involvement of TME was limited, resulting in incomplete ALDH profiling.Bearing this in mind, currently emerging evidence in PCa suggests the dominant isoforms are ALDH1A1,1A2, 1A3, 3A1 and 7A1. The expression and function have been demonstrated using a number of different 2D and 3D cancer models as well as clinical samples. Further investigations of these isoforms are required in order to fully validate their potential as biomarkers or targets for therapeutic intervention. Such investigations should take better account on our choices of models as argued by Maitland in accompanying review[224]in this thematic issue. As discussed in this review, ALDH enzymes also play a functional role in CSC populations,in the context of the TME. This synergy will be important in future studies to dissect whether ALDH expression leads to drug resistance via direct or indirect mechanisms. Underpinning the role of the RA signalling pathways, and the glycolytic biochemical pathways associated with the Warburg effect form part of both a regulatory network and a vicious cycle of tumour aggressiveness. The TME no doubt plays a critical role in exerting this selective pressure on ALDH expression and function, and hence should be more carefully considered in unravelling the cellular roles for specific ALDH isoforms. In this regard, use of siRNA, CRISPR and the development of highly specific small molecules to probe ALDH function will enable us more quickly ascertain the importance of specific ALDHs.
DECLARATIONS
Acknowledgments
We wish to acknowledge Prostate Cancer UK (RIA15-ST2-022 & PhD grant S12-027) forfinancial support and sponsorship, and Yaqeen Sawalha for producingfigures for this manuscript.
Author’s contributions
Manuscript writting and revision: Ibrahim AIM, Sadiq M, Frame FM, Maitland NJ, Pors K
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Con flicts of interest
The authors declare there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2018.
 Journal of Cancer Metastasis and Treatment2018年8期
Journal of Cancer Metastasis and Treatment2018年8期
- Journal of Cancer Metastasis and Treatment的其它文章
- AUTHOR INSTRUCTIONS
- Unmasking tumor heterogeneity and clonal evolution by single-cell analysis
- Centrosome aberrations and chromosome instability contribute to tumorigenesis and intratumor heterogeneity
- Increased ARF6 activation correlates with HGF stimulation in non-invasive prostate cancer cells
- Genomic heterogeneity meets cellular energetics:crosstalk between the mitochondria and the cell cycle
- Extracellular control of chromosomal instability and maintenance of intra-tumoral heterogeneity