
Cryptic NUP214-ABL1 fusion with complex karyotype, episomes and intra-tumor genetic heterogeneity in a T-cell lymphoblastic lymphoma
2018-11-20 12:25:14MoneebOthmanBeateGrygalewiczAgnieszkaKokowskaLeniakJoanaMeloIsabelCarreiraThomasLiehr
Moneeb A.K Othman, Beate Grygalewicz, Agnieszka Kołkowska-Leśniak, Joana B. Melo, Isabel M.Carreira, Thomas Liehr
1Jena University Hospital, Friedrich Schiller University, Institute of Human Genetics, Jena D-07740, Germany.
2Cytogenetic Laboratory, Maria Sklodowska-Curie Memorial Cancer Centre and Institute, Warsaw 02-781, Poland.
3Department of Lymphatic Diseases, Institute of Hematology and Transfusion, Warsaw 02-776, Poland.
4Laboratory of Cytogenetics and Genomics, Faculty of Medicine, University of Coimbra, Coimbra 3000-354, Portugal.
5Centro de Investigacao em Meio Ambiente, Genetica e Oncobiologia (CIMAGO), Coimbra 3001-301, Portugal.
Abstract T-lymphoblastic lymphoma (T-LBL) is a rare and aggressive form of non-Hodgkin’s lymphoma and little is known about their molecular background. However, complex karyotypes were already related to this group of malignancy and associated with poor outcome. Here, we describe a 17-year-old female being diagnosed with T-LBL and a normal karyotype after standard G-banding with trypsin-Giemsa (GTG)-banding. However, further analyses including highresolution molecular approaches, array-comparative genomic hybridization (aCGH), multiplex ligation-dependent probe amplification, fluorescence in situ hybridization and multicolor chromosome banding revealed a cryptic complex karyotype, NUP214-ABL1 gene fusion, episomes and intra-tumor genetic heterogeneity. In addition, homozygous loss of CDKN2A, as well as amplification of oncogene TLX1 (HOX11) were detected. Actually, NUP214-ABL1 fusion gene replicated autonomously in this case as episomes. Overall, highly amplification of NUP214-ABL1 fusion gene defines possibly a new subgroup of T-LBL patients which accordingly could benefit from treatment with tyrosine kinase inhibitors. As episomes are missed in standard karyotyping aCGH should be performed routinely in T-LBL to possibly detect more of such cases.
Keywords: T-cell lymphoblastic lymphoma, NUP214-ABL1 fusion, complex karyotype, episomes, intra-tumor genetic heterogeneity, molecular cytogenetics, array comparative genomic hybridization
INTRODUCTION
Lymphoblastic lymphoma (LBL) is a rare and aggressive form of non-Hodgkin’s lymphoma (NHL). LBL develops from immature B cells committed to the B- (B-LBL) or T-cell lineage (T-LBL). LBL is morphologically indistinguishable from acute lymphoblastic leukemia (ALL) and 90% of it have a T-cell phenotype. LBL also accounts for approximately 2% of all NHL cases and occur in adult, children and adolescent, with a male predominance (three time more male are affected)[1-2].
Chromosomal abnormalities in T-LBL are not well defined and cytogenetic data in T-LBL is limited. However, a few published cytogenetic studies revealed that typical chromosomal aberrations identified in T-cell ALL (T-ALL) are also present in T-LBL. These include translocations of T-cell receptor (TCR) gene to genes encoding transcription factors such asTAL1,TLX1,LMO2, andLYL1. In particular, the translocation t(9;17)(q34;q22~23) is typically found in T-LBL[1-4]. However, no single recurrent and typical genetic alteration for T-LBL could be identified. This is in contrast to other malignancies like translocation ofALKgene in anaplastic large cell lymphoma,MYCgene in Burkitt lymphoma orBCL2gene in follicular lymphoma.
Here we present the comprehensive analysis of a T-LBL case with a normal karyotype, according to standard G-banding with trypsin-Giemsa (GTG)-banding, using high resolution molecular methods, identifying also some intra-tumor genetic heterogeneity besides unusual acquired genetic alterations. Also here we reportNUP214-ABL1gene fusion in this patient, which appears cryptic due to its localization in episomes.
CASE REPORT
A seventeen-year-old female patient, who was initially diagnosed in South Africa with T-ALL, presented in the clinic in Poland with abdominal pain, accompanied by diarrhea and vomiting; she was here initially treated only symptomatically. A few days before, a blood test already revealed hyperleukocytosis (589 × 109/l)with presence of 94% lymphoblasts in blood smear, hemoglobin 8.5 g/dl, and platelet count 53 × 109/l. Bone marrow findings were: hypercellularity with 95% lymphoblasts, lack of megakaryocytes and Periodic-Acid-Schiff (PAS) staining identified in 70% of the blasts thick grains (data not shown). Ultrasound of abdomen showed enlargement of the spleen to 152 mm, and presence of fluid in the lower pelvis. Cervical lymph nodes were bilaterally enlarged with diameters of 3-4 cm, and small submandibular nodes were bilaterally enlarged to 2 cm in diameter.
Cytogenetic and immunophenotypic analyses were done. The latter characterized a T-LBL due to high expression of CD45 (100%), CD2 (96.6%), CD4 (97.3%), CD8 (90%), CD7 (77.1%), CD5 (76.0%), sCD3 (71.2%),CD1a (70.0%) and the lack of TdT, CD19, CD34 and CD38.
Banding cytogenetic analyses were done in unstimulated bone marrow cells according to standard procedures[5]from the material taken at initial diagnoses. A total of 20 metaphases were available and analyzed on a banding resolution of 300 bands per haploid karyotype, revealing a normal female karyotype. Molecular diagnostic polymerase chain reaction (PCR)-based tests for presence of gene fusionsBCR/ABL (p190 and p210),TCF3/PBX1,MLL/AF4and SIL/TAL1 were negative (results not shown).
Also genomic DNA isolated from cellsfixed in acetic acid-methanol (1:3) was subjected to array-comparative genomic hybridization (aCGH) as well as the multiplex ligation probe amplification (MLPA) studies, as previously reported[6], Finally, fluorescence in situ hybridization (FISH) was done[6-8], revealing a highly complex karyotype [Figure 1 and Table 1] with gene-amplification due to episomes (abbreviated here as epi), which can be reported as:
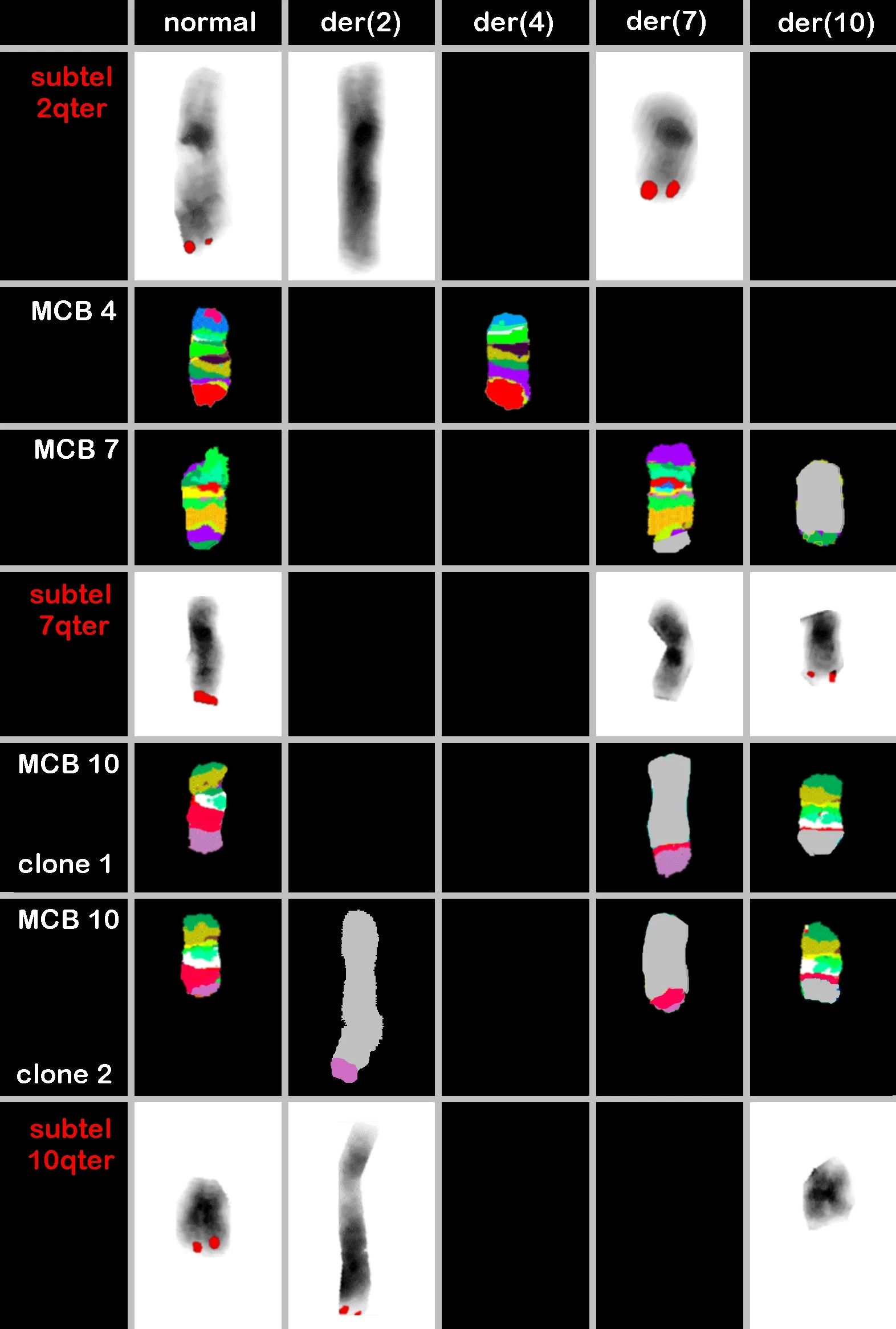
Figure 1. Result of multicolor banding (MCB) probesets for chromosomes 4, 7 and 10 are shown. MCB 10 showed the founder clone and subclone. Locus-specific probes (LSPs) for chromosomes 2, 7 and 10 characterized the breakpoints in 2q37.3, 7p34, 10q24.3 and 10q25.1[Table 1]. The final karyotype after application of all approaches is summarized in the text. der = derivative chromosome
In the FISH-studies done here, between 15 and 25 metaphases were evaluated per applied probe-set, thus in thefinal karyotype overall percentages are given for the observed clones.
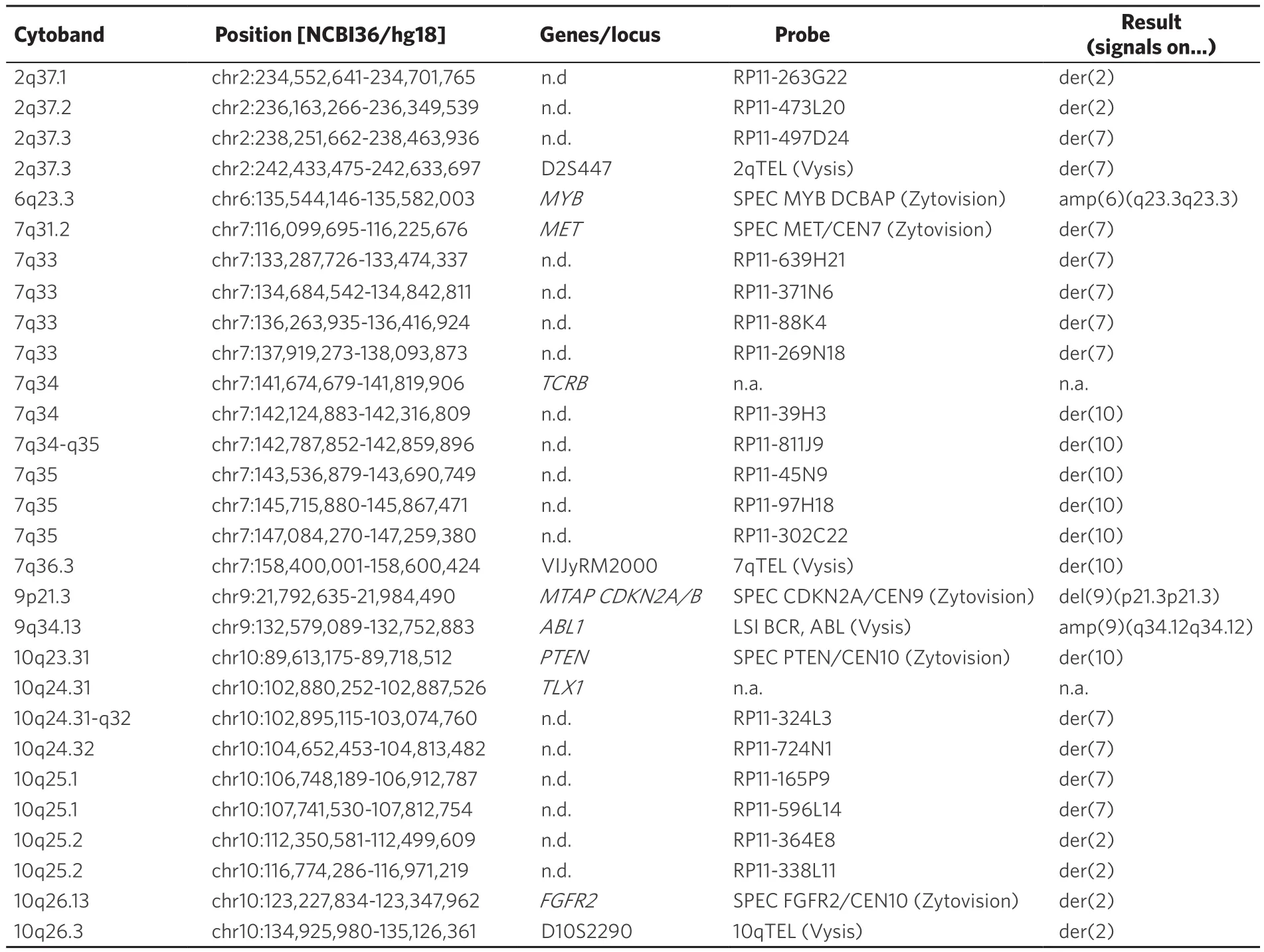
Table 1. Locus specific probes used for FISH together with their location according to genome browser version NCBI/hg18;this version was used here as some here applied FISH-probes are no longer available in newer genome browser versions.Results obtained are presented using standard (gene) abbreviations and such used according to the international system of cytogenomic nomenclature
NUP214-ABL1fusion could be deduced from aCGH data - the region being amplified ends on one side atNUP214- and on other side atABL1-gene - as the amplified region is present as episomes, which are circular,there must beNUP214-ABL1fusion.
The patient was treated according to the Polish Adult Leukemia Group (PALG) protocol, with induction therapy consisting of prednisone, daunorubicin, vincristine and PEG-L-asparaginase. No remission was achieved and the patient was re-treated according to fludarabine, cytarabine, and mitoxantrone (FLAM)with consolidation course (metrotrexate, cyclophosphamide and PEG-L-asparaginase) and maintenance treatment. After ten months, the patient relapsed and was now treated according to Hyper-CVAD protocol(cyclophosphamide, vincristine, doxorubicin/Adriamycin and dexamethasone). Still, one month later the patient unfortunately died.
DISCUSSION
Recurrent acquired genetic lesions play a key role in predicting and assessing risks, so are the treatment protocols to be applied. Still, little is known about the copy number alterations (CNAs) accompanying structural abnormalities in T-LBL, such as theNUP214-ABL1fusion gene.ABL1fusion proteins are sensitive to tyrosine kinase inhibitors, which potentially can be included in future treatment strategy andNUP214is a component of the nuclear pore complex, which mediates nucleocytoplasmic transport.NUP214is widely expressed and is involved in the pathogenesis of acute myeloid leukaemia associated with the translocation t(6;9)(p23;q34) asDEK-NUP214fusion[9-11].
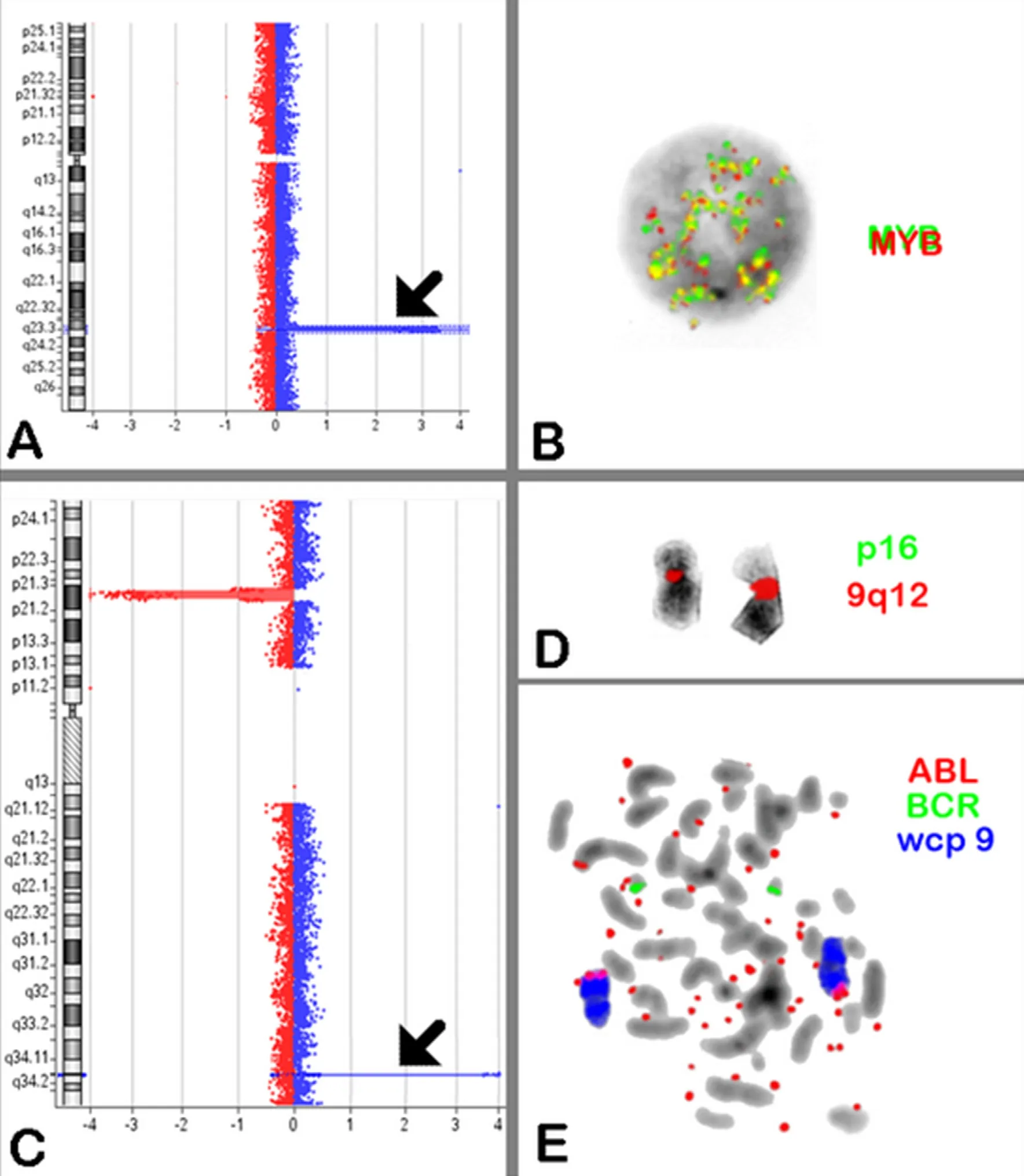
Figure 2. A: Array comparative genomic hybridization (aCGH) analysis of chromosome 6 revealed high level of 6q23.3 amplification containing MYB gene (arrow); B: MYB Dual Color Break Apart Probe was applied and showed high level of amplification more than 20 copies/per cell; C:aCGH analysis of chromosome 9 revealed biallelic deletion of CDKN2A at 9p21.3 and high level of 9q34 amplification contains ABL1 and NUP214(arrow); D: FISH confirmed the homozygous deletion of CDKN2A in metaphase; E: BCR, ABL Dual Color Probe was applied and showed variable number of episomes (20-30) in spread metaphases. aCGH: array-comparative genomic hybridization; FISH: fluorescence in situ hybridization;wcp: whole chromosome paint
To the best of our knowledge, a crypticNUP214-ABL1fusion yet has only been identified in 6% of individuals with T-ALL and is the second most prevalent fusion gene involvingABL1[12-15]. Here we report this for thefirst time in a T-LBL case, and even detected it as a high level amplification; most probably after inversion,duplication or translocation, gene fusion, circularization and amplification happened. AsABL1is one of the best targetable tyrosine kinases, identification ofABL1gene fusion is clinically important, as patients maypotentially benefit from tyrosine kinase inhibitors[16-17].
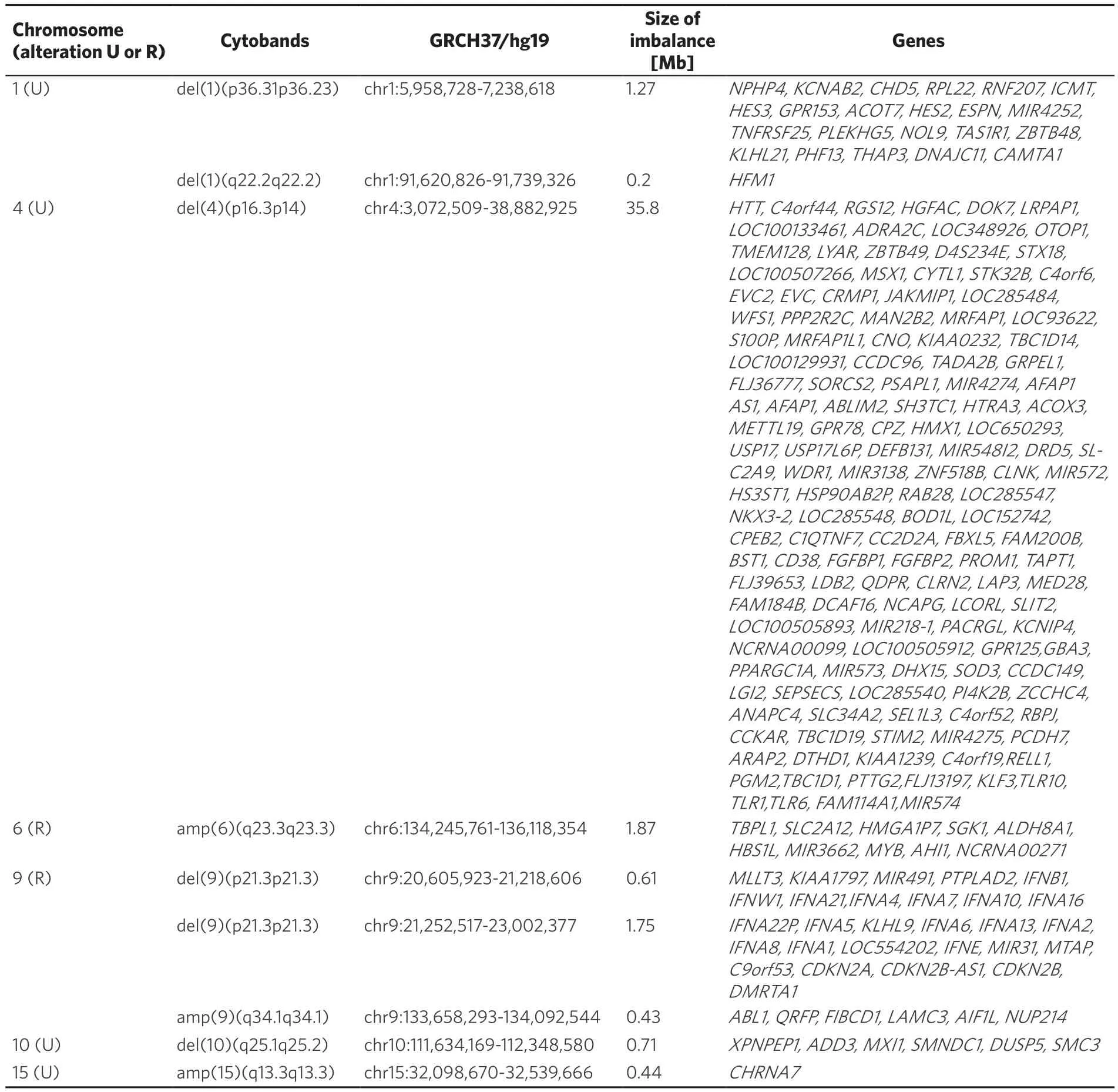
Table 2. Summary of CNAs detected by aCGH. Recurrent (R) and unique (U) acquired CNAs are correspondingly highlighted in first column. Results obtained are presented using standard (gene) abbreviations and such used according to the international system of cytogenomic nomenclature
Episomes are submicroscopic, circular and large acentric DNA fragments that can replicate autonomously.One of the common formation-mechanisms for extrachromosomal elements in cancer cells is episome-replication and unequal segregation during cell division, resultingfinally in an increase of copy numbers. Still they that are invisible in banding cytogenetics; this is because episomes are composed of only several hundred kilobases of amplified oncogenes and/or drug-resistance genes, and thus are too small to be visualized by light-microscopy[18-20]. Of interest, we detected a variable number of episomes (20-30) in different cells.However, we suggest that c-MYBis also present on the same episomes [Figure 2]; due to lack of material we could not confirm this by FISH.
Additionally, recurrent acquired CNAs in different chromosomal regions were also identified besides unique ones for this case [Table 2]. Taken together, the genomic abnormalities in T-ALL and T-LBL are so similar that they could be considered as identical diseases in the future[1,4,12,14,15,21-24].
As shown in our case,NUP214-ABL1is accompanied with loss of cyclin-dependent kinase inhibitor 2A(CDKN2A), which encodes the tumorsuppressors p16INK4A and p14ARF, and affects cell cycle progression.CDKN2Agene deletion can be detected at initial diagnosis or acquired at relapse, suggesting thatCDKN2Agene deletion is a secondary genetic event and associated with chromosomal rearrangements. This may as a result lead to the aberrant expression of a diverse group of T-cell-specific transcription factors, which again can function as oncogenes, such asTLX1andTLX3[2,21,25]. The translocation t(7;10)(q34;q24), resulting from theTRB/TLX1fusion gene, has been reported in several studies, and is present in 5% of pediatric and 30% of adult with T-cell ALL[25-28].
Overall, in the present T-LBL case we identified substantial intra-tumor genetic heterogeneity and complexity. The founder clone hasTRB/TLX1fusion gene and the subclone hasTRB/TLX1fusion gene plus complex karyotype involving three-way translocation t(2;7;10)(2q37.3;7q34;10q25.1), further developing into a more complex subclone. Interestingly, the breakpoints at 2q37.3, 7q34, 10q24.3 and 10q25.1 were not previously reported in T-LBL[29]. Thus, this data provides genetic support for a multi-step pathogenesis: deletion of a tumor-suppressor gene (CDKN2A), deregulated expression of a transcription factorTLX1and most likely overexpression of a constitutively activated tyrosine kinase (NUP214-ABL1) and oncogenec-MYBdue to episome amplification and the unique phenotypes of the T-LBL case mentioned above.
To conclude, this study demonstrates the power of high resolution molecular approaches. It may be considered that the use of such approaches is the most efficient and future standard method for screeningABL1alteration. Particularly in T-LBL patients this may be advantageous, asABL1modulates T-cell development and plays a role in cytoskeletal remodeling processes in T-cells. Besides, the intra-tumor genetic heterogeneity in cancer has important implications for reservoirs of cells involved in progression of disease and drug resistance therapy. AsNUP214-ABL1fusion is sensitive to the tyrosine kinase inhibitor, this suggests that new therapeutic approaches in T-LBL may improve outcome and/or decrease treatment-related morbidity.
DECLARATIONS
Authors’ contributions
Did the FISH-studies and drafted the paper: Othman MAK
Performed array comparative genomic hybridization (aCGH) analyses and interpretation: Melo JB, Carreira IM, Othman MAK
Provided T-LBL-case including clinical and banding cytogenetic data: Grygalewicz B, Kołkowska-Leśniak A Planned and organized the study and didfinal drafting of the paper: Liehr T
All authors read and approved the paper.
Availability of data and materials
All data is provided in this article. Also the patient was mentioned previously in[30]as P61.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no con flicts of interest.
Ethical approval and consent to participate
The present study was approved by the Ethical Board at the Friedrich Schiller University (Jena, Germany;approval No., 1105-04/03). Consent to participate of the parent of the patient studied here is available on request from the authors of this paper.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2018.
 Journal of Cancer Metastasis and Treatment2018年9期
Journal of Cancer Metastasis and Treatment2018年9期
- Journal of Cancer Metastasis and Treatment的其它文章
- AUTHOR INSTRUCTIONS
- Two sides to colon cancer: mice mimic human anatomical region disparity in colon cancer development and progression
- NSAID celecoxib: a potent mitochondrial pro-oxidant cytotoxic agent sensitizing metastatic cancers and cancer stem cells to chemotherapy
- Introduction to the special issue on reviews of gastric cancer metastasis and treatment
- Pharmacogenetics and cancer management