
基于E6/E7基因检测高危型人乳头瘤病毒感染新方法的构建*
2018-11-08 07:24:50谢旺凯吕诗卉薛向阳陈向敏
检验医学与临床 2018年21期
王 俭,谢旺凯,李 婷,吕诗卉,倪 超,薛向阳,陈向敏,陈 俊△
(温州医科大学:1.第一临床医学院;2.第二临床医学院;3.基础医学院;4.检验医学院/生命科学院,浙江温州 325035)
宫颈癌是一种严重危害女性健康的恶性肿瘤,位于女性恶性肿瘤的第二位[1]。研究已证实,几乎所有宫颈癌都是由人乳头瘤病毒(HPV)尤其是高危型 HPV感染引发,因此高危型HPV检测逐渐成为宫颈癌筛查的研究热点[2]。目前,HPV研究主要针对HPV基因组进行。HPV基因组可分为早期区、晚期区及上游调节区(URR)或长控制区(LCR)或非编码区(NCR)。早期区编码E6、E7、E1(E8)、E2 、E4 (E3)、E5等5~8个基因,晚期区编码L1和L2病毒结构蛋白[3]。目前国内外广泛采用HPV通用引物定位在HPVL1基因上进行检测[4]。但是在高危型HPV感染过程中,其基因会整合到宿主细胞基因组中,这是引发宫颈细胞癌变发生的前提[5]。而高危型HPV DNA的整合过程往往会发生基因断裂缺失,如E1/E2调控区和L1/L2衣壳蛋白基因的缺失,因此基于L1基因的检测存在一定的漏诊现象。然而,研究表明E6和E7基因在整合过程中却可以保留完整,不易发生缺失,因此,E6/E7基因被认为是检测HPV最为理想的靶点[6]。基于以上现象,本研究针对HPV E6/E7基因,设计我国感染率较高的7种高危型HPV特异性引物,包括HPV16、18、31、33、51、52、58,建立一种成本低廉、效率更高的检测宫颈肿瘤、脱落细胞等组织高危型HPV感染的多重聚合酶链反应(PCR)检测方法,并与现在临床上主流的流式荧光杂交法进行比较,评价其临床意义。
1 材料与方法
1.1标本来源
1.1.1宫颈肿瘤组织标本 68份宫颈肿瘤组织标本采自温州医科大学附属第一医院妇产科68例手术患者,并经病理检查确诊,其中宫颈上皮内瘤变(CIN) 1级12例,CIN 2级19例,CIN 3级19例,宫颈鳞癌12例,宫颈腺癌2例,正常宫颈(内膜透明细胞癌)4例,标本离体后保存于液氮中。患者年龄25~69岁,平均(42.4±1.1)岁。所有标本收集均征得患者及或家属的同意,并签署知情同意书。
1.1.2宫颈脱落细胞标本 150份宫颈脱落细胞标本均取自温州医科大学附属医院妇科门诊患者。患者年龄18~78岁,平均(43.1±11.6)岁。所有标本收集均征得患者及或家属的同意,并签署知情同意书。HPV16阳性的Siha细胞和HPV18阳性的Hela细胞,购自美国ATCC,并由实验室保存。
1.2DNA提取 宫颈肿瘤组织标本每份切取50 g病变区组织,碾碎后加入50 μL磷酸盐缓冲液(PBS)中制成悬液备用;Siha、Hela细胞贴壁培养至长满50 mL培养瓶后,也制成悬液备用。制备的悬液采用DNA提取试剂盒[TIANGEN TIANamp Genomic DNA Kit(DP304)]提取DNA。将DNA模板统一稀释至100 ng/μL。宫颈脱落细胞溶于细胞保存液中,取2 mL细胞溶液10 000 r/min离心1 min后去上清液,采用DNA提取试剂盒提取DNA。
1.3引物
1.3.1引物设计 在分析同源性的基础上,针对我国流行的主要高危型HPV16、18、31、33、51、52、58及相应HPV基因组的E6/E7基因,设计这7种HPV型别的引物。由北京六合华大基因科技有限公司合成[7]。引物序列见表1。将合成的引物稀释成10 μmol/L,-20 ℃保存,待用。
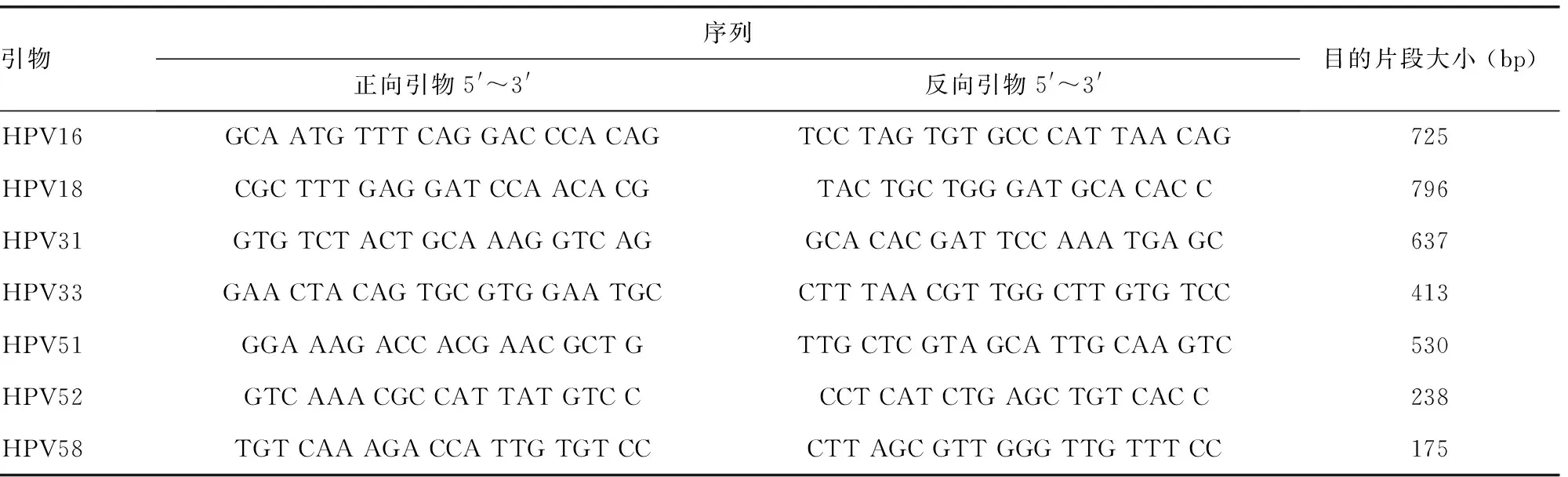
表1 各型别HPV特异性引物序列
1.3.2引物特异性验证 为验证相应HPV型别引物特异性,以明确HPV分型的宫颈肿瘤组织标本和细胞株的DNA为模板,引物FP 10 μmol/L,引物RP 10 μmol/L,Taq Mix 12.5 μL[TIANGEN 2×Taq PCR MasterMix(KT201)]配制成25 μL反应体系。退火温度为63 ℃(16、18、31、33、51、58型),65 ℃(52型)。热循环条件设置如下:94 ℃预变性5 min;94 ℃变性30 s;退火30 s;72 ℃延伸1 min,35个循环;最后72 ℃后延伸5 min。在Bio-Rad S1000 PCR仪上进行反应。PCR产物琼脂糖凝胶电泳,切胶回收,基因测序,并与已公布的各型别HPV基因序列比较。
1.3.3引物敏感性验证 为分析特异性引物敏感性, 以10.00、1.00、0.10、0.01 ng不同HPV型别阳性的DNA作为模板进行试验,PCR产物进行琼脂糖凝胶电泳,并进行敏感性分析。
1.4E6/E7多重PCR检测方法的构建 以7种明确HPV分型的宫颈肿瘤组织标本为DNA模板,将HPV16、18、31、33、51、52、58型的特异性引物按浓度1∶1∶1∶1∶1∶1∶1的比例混合加入4 μL,RNase-free去离子水1 μL,Taq Mix 7.5 μL;Q-Solution+1.5 μL[QIAGEN Multiplex PCR kit(Cat.No.206143)]配制成15 μL体系。热循环条件设置如下:95 ℃预变性15 min;94 ℃变性30 s;63 ℃退火90 s;72 ℃延伸45 s,35个循环;最后68 ℃后延伸15 min。PCR产物进行琼脂糖凝胶电泳,对比各型别HPV的单一PCR产物电泳图谱以确定多重PCR方法的特异性。在此基础上,以10.00、1.00、0.10、0.01 ng不同HPV型别阳性的DNA作为模板进行试验,PCR产物进行琼脂糖凝胶电泳,并进行敏感性分析。同时,为避免实验存在的偶然性,相隔一定量的时间后,同样以新建立方法时所使用的7种高危型别阳性标本DNA作为模板,在相同条件下以同样的参数进行实验,验证多重PCR方法的稳定性。
1.5宫颈肿瘤组织标本HPV感染型别检测 对68份宫颈肿瘤组织标本采用所构建的多重PCR方法进行检测。同时对相同标本采用流式荧光杂交法进行平行检测,记录并比较分析结果。
1.6宫颈脱落细胞标本检测 使用新建立的多重PCR方法对150份宫颈脱落细胞标本进行HPV感染情况检测,同时对相同的标本采用流式荧光杂交法进行平行检测,记录并比较分析结果。
1.7统计学处理 采用SPSS 21.0统计软件进行数据处理及统计分析,计数资料以例数或率表示,组间比较采用χ2检验,P<0.05为差异有统计学意义。
2 结 果
2.1引物设计结果 引物特异性实验表明7种高危型HPVE6/E7引物(HPV16、18、31、33、51、52、58)均具有特异性,见图1。同时敏感性分析实验显示:HPV16、18、33型DNA 模板量低至1 ng仍能扩增特异目的片段,HPV31型DNA模板量低至10 ng仍能扩增特异目的片段,HPV51型DNA模板量低至0.1 ng仍能扩增特异目的片段,HPV52、58型DNA模板量低至0.01 ng仍能扩增特异目的片段。
2.2E6/E7多重PCR检测方法的建立 对已经明确了HPV分型的宫颈肿瘤标本和细胞株所提取的DNA进行多重PCR扩增,结果显示出现单一条带,与使用单一型别引物进行PCR的结果一致,提示所建立的多重PCR可特异检测这7种HPV亚型,见图2。且敏感性分析实验显示:HPV16、18、31、33、58型DNA模板量低至10 ng仍能扩增特异目的片段,HPV51和52型DNA模板量低至0.1 ng仍能扩增特异目的片段。
2.3宫颈肿瘤组织HPV检测结果 以新建立的多重PCR方法检测68份宫颈肿瘤组织标本,见图3,根据电泳条带判断68份宫颈肿瘤组织标本HPV感染情况,检测出HPV阳性标本48份(70.59%),其中HPV16阳性36份(52.94%),HPV18阳性3份(4.41%),HPV31阳性4份(5.88%),HPV33阳性7份(10.29%),HPV51阳性2份(2.94%),HPV58阳性3份(4.41%),多重感染6份(8.82%)。高于流式荧光杂交法检测所显示39.71%的感染率,2种方法检测结果比较,差异有统计学意义(χ2=11.43,P<0.05)。

注:M为D2000 DNA标记物;1为HPV16阳性标本;2为HPV18阳性标本;3为HPV31阳性标本;4为HPV33阳性标本;5为 HPV51阳性标本;6为HPV52阳性标本;7为 HPV58阳性标本
图1所设计的7种高危型别HPV E6/E7引物的特异性分析

注:A为HPV阳性标准条带;B为HPV 7种亚型引物特异性分析;M1为D2000 DNA 标记物;M2为阳性标准条带;1为HPV16阳性标本;2为HPV18阳性标本;3为 HPV31阳性标本;4为HPV33阳性标本;5为HPV51阳性标本;6为HPV52阳性标本;7为HPV58阳性标本
图2多重PCR法电泳结果分析
2.4宫颈脱落细胞标本检测结果 150份宫颈脱落细胞标本用新建立的多重PCR方法及流式荧光杂交法检测的结果显示7种高危型别(HPV16、18、31、33、51、52、58)检出率比较,差异无统计学意义(χ2=3.68,P>0.05),见表2。
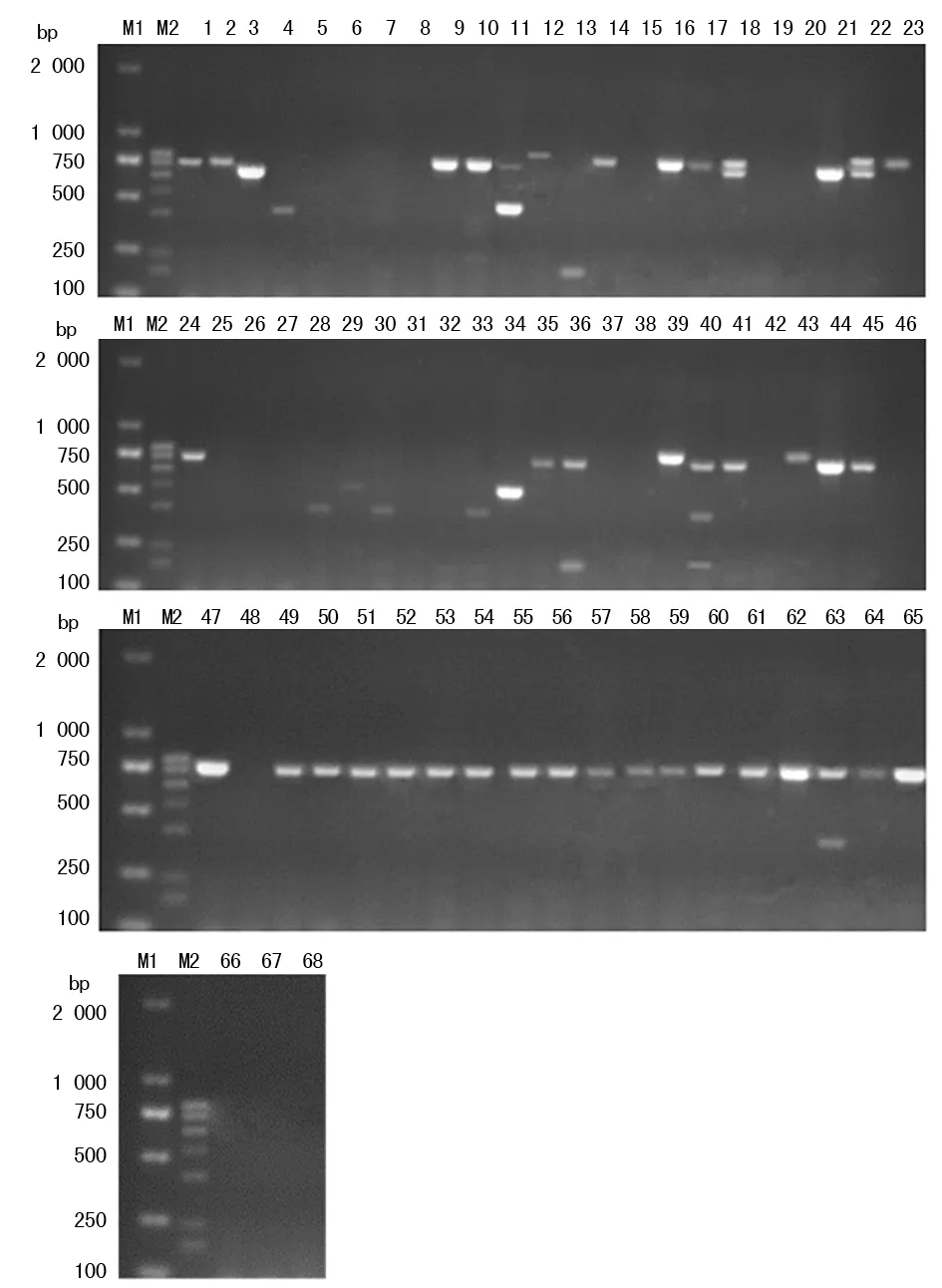
注:M1为 D2000 DNA标记物;M2为阳性标准条带;1~68为宫颈肿瘤组织标本
图3 68份宫颈肿瘤组织多重PCR检测结果分析

表2 150份宫颈脱落细胞不同检测方法的HPV检测结果(n=150)
3 讨 论
高危型HPV持续感染是导致宫颈癌发生的重要原因。越来越多的研究表明HPV基因组检测对宫颈病变预测和术后疗效判断有利,可以避免不必要的诊断和治疗。因此,国内外采用了很多种方法对HPV感染进行检测,应用于临床宫颈癌筛查和防治。
现在临床上采用较多的是基于L1衣壳蛋白基因的HPV检测方法,如流式荧光杂交法[8]。但由于在HPV感染过程中,病毒 DNA通常会与宿主染色体整合,会改变整合位点及邻近宿主基因的表达,使部分HPV L1基因组发生断裂丢失,最终导致HPV L1衣壳蛋白也表达缺失,所以基于L1基因序列的HPV检测方法容易出现假阴性的结果,这也限制了其在临床上的使用[9]。而早期蛋白基因E6/E7在肿瘤的发生过程中持续存在,且其表达的E6/E7早期蛋白在宫颈癌变过程中起着重要作用。恶性肿瘤病变时,HPV DNA整合到宿主DNA当中,通常导致E2基因中断从而使E6/E7基因表达增强[10]。E6/E7蛋白分别抑制P53、Rb基因的活性,激活人细胞端粒酶的转录,引起细胞异常分化,导致正常细胞永生化,最终形成肿瘤[11]。因此,对HPV E6/E7基因进行检测,能从根源上对宫颈病变进行诊断和预防,是判断HPV感染的更好选择。然而,临床上针对E6/E7的检测方法不多,尤其是多重PCR的检测方法几乎没有。为此本研究主要构建了基于E6/E7的多重PCR方法检测HPV的感染情况,在对HPV进行同源性分析后证实各HPV亚型的E6/E7基因具有特异的保守片段,据此选择设计并验证了中国人群感染率较高的7种高危型别HPV E6/E7的特异性引物,优化条件后建立了一种基于E6/E7基因的HPV多重PCR分型检测方法。对已明确型别的宫颈癌组织标本及宫颈细胞的检测阳性标本出现条带,而阴性标本未出现条带证明其特异性好,而且实验还验证了该多重PCR技术检测灵敏度能达到10.00 ng,技术稳定性良好,满足临床标本检测方法的要求。
68份宫颈肿瘤组织标本的HPV检测结果显示,所构建的基于E6/E7多重PCR检测阳性率明显高于基于L1基因的流式荧光杂交法检测,差异有统计学意义(P<0.05)。而对门诊采集的150份宫颈脱落细胞标本检测结果显示7种高危型别检出率与流式荧光杂交法检出率并无差异,考虑到门诊患者高危型别感染较少,样本数不够大,有待进一步验证。
临床上使用的基于L1基因的流式荧光杂交法检测费用较高,操作复杂,需要大型仪器设备支持,不利于在乡镇卫生院开展。而本研究所建立的基于E6/E7多重PCR法不仅能够一次检测HPV16、18、31、33、51、52和58 7种高危型别,而且具有操作简便、成本低廉和所需仪器较少且较易获得等优势,同时研究结果也已表明两种方法检出率高度相近。因此,基于E6/E7多重PCR法在应用中能极大弥补流式荧光杂交法在成本和技术等方面的不足,同时可以减少多次检测带来的交叉污染及高成本的问题,适合在临床大面积开展患者高危型HPV感染的早期筛查。
猜你喜欢
青少年科技博览(中学版)(2023年1期)2023-03-17 00:44:34
黄河·黄土·黄种人(华夏文明)(2021年6期)2021-09-28 02:14:08
现代仪器与医疗(2021年1期)2021-06-09 05:53:54
中华养生保健(2020年7期)2020-11-16 01:14:16
中国生殖健康(2019年3期)2019-02-01 06:12:22
中国生殖健康(2019年3期)2019-02-01 06:12:08
中国生殖健康(2019年9期)2019-01-07 01:18:48
健康必读·下旬刊(2018年4期)2018-06-04 08:43:16
中国卫生标准管理(2015年25期)2016-01-14 09:29:15
中国医药科学(2015年15期)2015-02-27 12:32:36
