
2-氨基噻唑类化合物抗肿瘤机制的研究现状
2018-11-06 07:43付竹孙文超栾相成高锦欣张志华
锦州医科大学学报 2018年5期
付竹,孙文超,栾相成,高锦欣,张志华
(辽宁工业大学化学与环境工程学院,辽宁 锦州 121001)
2-氨基噻唑(2-AT)作为苯酚、儿茶酚的电子等排体[1]。它主要由一个五元噻唑杂环构成,环上的1、3位含有硫、氮两种元素[2]。近年来,人们逐渐发现了2-氨基噻唑衍生物可做为治疗癌症和细胞增殖性疾病的治疗剂,其作用机制主要体现在以下几个方面。
1 PGE2表达抑制剂
25%的癌症的发生过程与炎症有关,前列腺素E2(PGE2)是调节疼痛和炎症的关键因素,在结肠、肺癌、乳腺癌、头颈癌等癌细胞中过度表达。高剂量的非甾体抗炎药(NSAIDs)会导致大肠癌肿瘤衰退,主要原因就是这些药物能减少PGE2的水平[3],但是,同时高剂量和长期使用的非甾体类抗炎药,可引发包括胃肠不耐受、心脏病发作和血液凝固等诸多副作用,因此,合成一种降低PGE2表达水平的替代性抗癌药物很有意义。Breland Smith[4]合成36个2-氨基噻唑类似物并测定它们对PGE2还原性,化合物1体外对PEG2的还原活性最强(EC50=90 nM),但对COX-2抑制性IC50值>5M。
2 Hec1/Nek2抑制剂
Hec1(癌症高表达蛋白)是外层着丝粒纺锤体检查点调节器重要的组成之一,在有丝分裂过程中控制着着丝粒-微管附着,染色体排列,并能维持有丝分裂阻滞缺陷蛋白2(MAD2),防止细胞过早进入有丝分裂后期[5]。Hec1的激活受Nek2调节,Nek2是细胞周期调控蛋白激酶Ⅰ(NIMA)家族成员之一,具有丝氨酸/苏氨酸蛋白激酶活性。Hec1 /Nek2和癌症的发生过程密切相关,多种人类癌症如NCI-60中均发现Hec1的过度表达,Hec1也是原发性乳腺癌和多发癌患者预后良好的标志[6]。Lee等[7]设计并合成了一系的4-芳基-N-芳基羰基-2-氨基噻唑化合物,可做为Hec1/Nek2抑制剂(见图1),化合物2在体外表现出低纳摩尔级的抗增殖活(IC50:16.3~42.7 nM),SD大鼠体内,高静脉AUC(64.9M/ h,2 mg/kg),并对异种移植人MDA-MB-231乳腺癌的小鼠体现出显著的体内抗肿瘤活性(T/C=32%,20 mg/kg,Iv)。C-M Hu[8]合成了INH衍生物3,它们可与Hec1蛋白394-408结构域的W395,L399和K400残基的相互作用结合,该区域紧邻Nek2的特异性结合区域,能有效阻断Nek2对Hec1 S165残基的磷酸化。INH衍生物对HeLa、MB468细胞显示纳摩尔范围的活性,并能有效抑制小鼠体内肿瘤的生长,没有明显的毒性。Yongxia Zhu[9]以INH为模版,合成了一种对K562肿瘤细胞有明显抑制作用的化合物4,用于慢性髓细胞白血病(chronic myelocytic leukemia,CML)潜在的治疗。免疫共沉淀结果表明4与INH的作用机制相似,干扰Nek2结合Hec1,并降低了Nek2、Hec1的表达量,诱导细胞G0/G1期静息和凋亡。

图1 PGE2及Hec1/Nek2抑制剂的结构
3 Chk1抑制剂
细胞周期检查点激酶1(Chk1)由人类基因组编码的一种丝氨酸/苏氨酸蛋白激酶,它主要参与细胞核内DNA复制及损伤修复等一系列过程。当DNA受到损伤时,Chk1被ATM和ATR磷酸化激活,造成细胞G2 期静息。同时,Chk1还能激活一系列修复基因的转录促进受损DNA进行修复,以保持基因组的完整性[10]。肿瘤细胞中由于P53基因突变,缺失G1期检查点,必须依赖Chk1检查点途径,才能对一些理化因素造成的基因损伤进行修复,进而存活[11]。研究发现,Chk1抑制剂(见图2)能明显增加DNA损伤性化疗药物的药效并避免抗药性的产生。Dudkin VY[12]等针对Chk1的ATP竞争结合位点合成了一系列吡啶氨基噻唑类化合物,其中含有乙二胺酰胺基的化合物体现出皮摩尔的抑制活性和较长的作用时间。化合物5与Chk1的晶体X-衍射结果显示,乙二胺酰胺基的酰基氧与lys38形成氢键,乙二胺酰胺基末端的氨基基团分别与Asn135、Asp148形成两摩尔氢键,解释了该类化合物高活性的原因。
该课题组还发现Chk1抑制剂处理后,H1299肿瘤细胞仍能逃避DNA损伤,进入细胞静息和有丝分裂,检查点逃避分析(checkpoint escape assay,CEA)表明胞内Chk1抑制剂活性的丧失可能与Cdk7活性有关。Cdk7是细胞周期依赖性激酶蛋家族的一员,其主要作用是维持细胞周期的转换,调节细胞周期进程。cdk7抑制剂可能导致细胞静息,使Chk1抑制剂介导检查点逃避分析失效。因此,在Chk1抑制剂的设计过程中还应注意考虑Cdk7活性、分子量和分子的表面极性和亲脂性等方面的因素。经过对2-氨基噻唑的结构修饰,化合物6及7显示出较好的CEA活性[13]。

图2 Chk1抑制剂的结构
4 KDR激酶抑制剂
VEGFR-2又称KDR或Flk- 1(kinase insert domain-containing receptor),具有调节淋巴管内皮细胞和血管内皮细胞,促进淋巴管和血管的生成等作用,存在于血管和淋巴管内皮等处,在多种癌细胞中VEGFR-2表达上调,是抗血管新生疗法的重要靶标之一[14]。Bilodeau MT等[15]以一种偶氮染料的结构为模版,进行结构改造,得到化合物8(见图3),它显示出较好的KDR酶活性和细胞毒性。随后又在8基础上结构改造,合成了其它N-(1,3-噻唑-2-基)吡啶-2-胺[16],9在表现出优异的体内活性同时能避免对hERG的抑制,延长QTc,具有良好的体内药动学。
Sahar M. Abou-Seria等[17]合成了一系列酞嗪衍生物,含2-氨基噻唑环的化合物10对VEGFR-2具有亚微摩尔级的抑制,IC50值为(0.40±0.0)μM。同时,体外抗肿瘤活性检测证明10是最有效的抗癌剂,对白血病、前列腺癌的GI50值(MG-MID)分别为3.51和5.15 μM。分子对接显示,N-H作为重要的氢键供体,与GLu885乙酰基形成氢键。同时,与蛋白之间存在四种芳烃相互作用,分别是酞嗪环和Leu840,哌嗪环与Phe1047,氨基噻唑环与Ile1044和Asp1046。

图3 KDR激酶抑制剂的结构
5 DNA复制抑制剂
纺锤菌素(netropsin,NT)是由链霉菌产生的一种碱性肽类抗生素,能选择性地与B型DNA小沟中的富含A-T碱基对的区域结合,使DNA由A型转变为B型,从而丧失模版的功能。Laila A. Abouzeid[18]基于NT的结构,合成了9种2-氨基噻唑类似物,11活性最好(IC50=38.3M)。课题组还模拟了这些化合物与β-DNA的结合模式,相关系数为R2=0.94,可为此类化合物进一步的结构修饰提供参考。
6 KPNB1抑制剂
亲核素β1基因蛋白(importin β1,KPNB1)参与细胞核转运蛋白的过程。癌症细胞中,KPNB1在转运磷酸化STAT3(pSTAT3)、细胞膜受体、含NLS序列的受体酪氨酸激酶(RTK)如ErbB2及表皮生长因子受体(EGFR)等的过程中发挥作用。这些受体进入细胞核后与细胞周期蛋白(cyclin D1),环氧合酶-2(COX-2),极光激酶A,原癌基因蛋白c-Myc,乳腺癌耐药蛋白(BCRP),STAT1及B-Myb等结合,促进细胞周期进程[19]。竞争性蛋白结合分析法显示[20],12与KPNB1具有强大的亲和力(Kd:20 nM)。Western blot法显示12能导致核内KPNB1、KPNA2、EGFR、ErbB2和STAT3含量的降低,确认了12对入核蛋白通路的抑制作用。
7 人含缬酪肽蛋白抑制剂
人含缬酪肽蛋白(VCP,又名P97),在多种生物中发挥作用。如ERAD(内质网相关降解)、有丝分裂完成后的核膜融合、高尔基重组、抑制凋亡、激活转录因子等。此外,VCP能与IκB作用,使之降解,从而激活NFκB通路[21]。Matthew G[22]发现2-苯胺基-4-芳基-1,3-噻唑对VCP有潜在活性,R1=4-OH及4-NH2的2-苯胺基-4-(4-氯苯基)-1,3-噻唑13、14活性最好(IC50分别为0.07 μM,0.08 μM)。
8 去乙酰酶抑制剂
组蛋白乙酰转移酶(HAT)和组蛋白去乙酰酶(HDAC)是影响表观遗传学活性的关键酶,利用这两种酶可对组蛋白氮端氨基酸残基进行乙酰化和去乙酰化,调节染色质的结构,进而调控基因转录。抑制HDAC的活性,能够引起细胞中乙酰化组蛋白的堆积,使p21、p53等基因的表达水平增加,达到抑制肿瘤细胞的增殖、诱导细胞分化和凋亡的目的。SampathKumar Anandan[23]等设计了噻唑环连接哌嗪环、哌嗪环上带有硫胺基的结构15(见图4),这些新的分子,有效地抑制宫颈癌Hela细胞中提取的HDAC酶,并对乳腺癌MCF7细胞具有抗增殖活性。
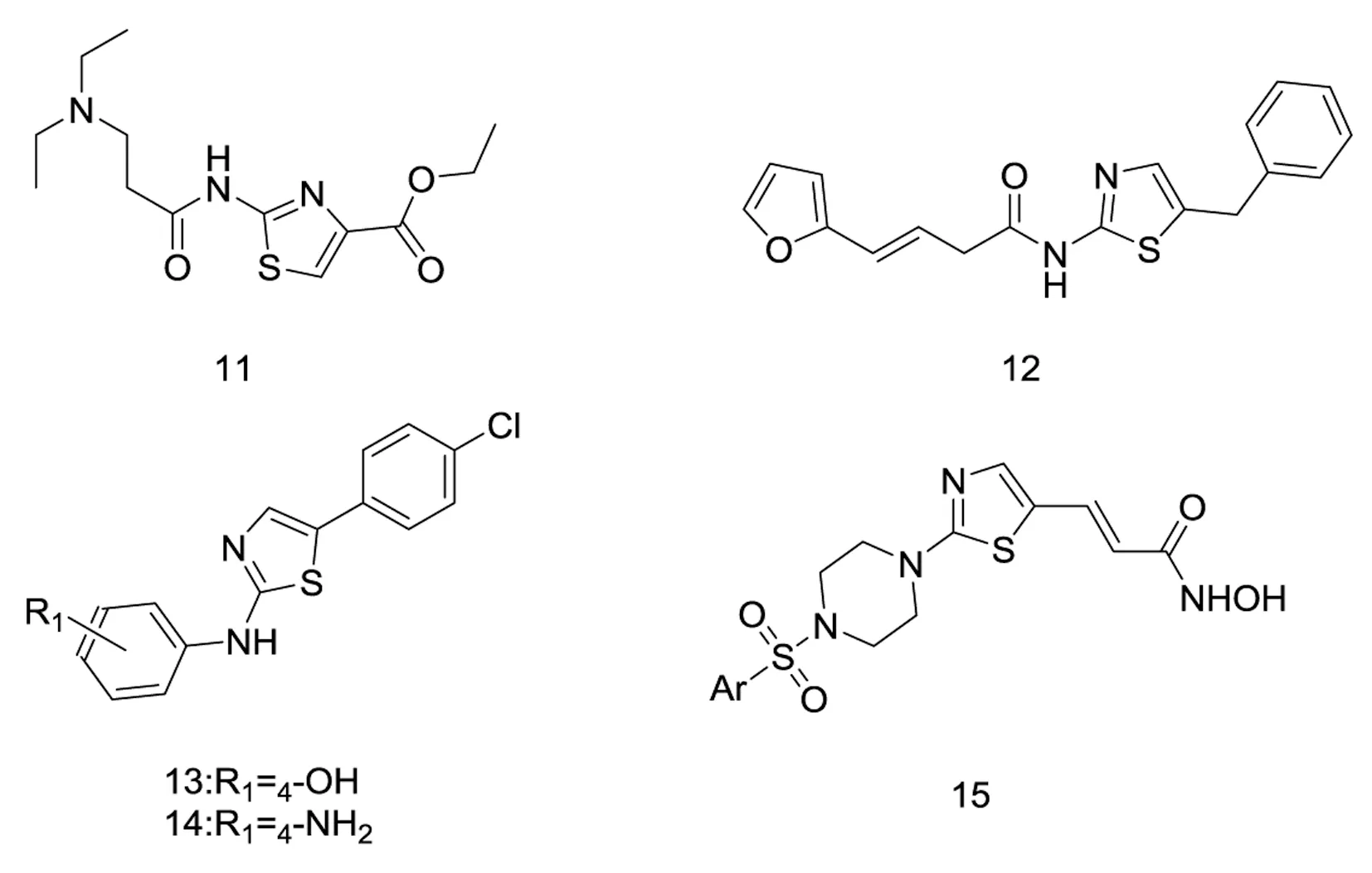
图4作用于其它靶点的2-氨基噻唑衍生物的结构
9 展望
在噻唑类环2位引入氨基方便噻唑同其它活性片段拼接,而且可改善化合物水溶性。其独特的结构决定了它很容易与靶标蛋白形成氢键、π-π堆积、静电和疏水等多种非共价键相互作用,因此,具有2-氨基噻唑环的化合物在医药领域尤其是抗肿瘤药物领域具有广泛的应用。随着对其结构关系及作用机制的不断研究,开发新型、高效、低毒的2-氨基噻唑类抗肿瘤药物正逐步成为研究热点。
猜你喜欢
化工与医药工程(2022年3期)2022-08-08
计算机系统应用(2022年4期)2022-05-10
天津医科大学学报(2021年4期)2021-08-21
计算机应用(2017年1期)2017-04-17
癌症进展(2015年3期)2015-12-18
郑州大学学报(工学版)(2015年1期)2015-03-24
火炸药学报(2014年5期)2014-03-20
郑州大学学报(理学版)(2014年4期)2014-03-01
浙江科技学院学报(2014年6期)2014-02-28
中国信息化·学术版(2013年5期)2013-10-09
