
结直肠癌的分子分型及精准医学时代展望
2018-10-22 01:36孙玉琳赵晓航
中华结直肠疾病电子杂志 2018年5期
孙玉琳 赵晓航
1990年,Fearon和Vogelstein提出了结直肠癌(colorectal cancer,CRC)癌变的多步骤、多阶段遗传演进模型。在这一模型中,最早出现的是正常结肠黏膜中抑癌基因APC(adenomatous polyposis coli)的失活突变,诱导癌前状态腺瘤的产生,随后是癌基因KRAS(kirsten rat sarcoma viral oncogene homolog)的激活突变,以及SMAD4(SMAD family member 4)和 TP53(tumor protein p53)等基因的依次突变,使得腺瘤逐渐发展为癌[1-2]。这一模型因此也被称作“腺瘤——癌序贯模型”(adenoma–carcinoma sequence model)。这些基因的累积突变通过调节细胞增殖、分化和凋亡通路从而驱动结直肠细胞的癌变,如Wnt-β-catenin、TGF-β、PI3K、TP53、EGFR和下游的RAS-MAPK通路等[2-3]。
“腺瘤——癌序贯模型”真的能够完全解释散发型CRC的癌变机理和临床病理特征吗?人们前期发现,某些患者的CRC肿瘤中不具有或仅存在一条上述驱动性通路的改变,但是却具有显著的染色体异常和/或表观遗传学改变[3],这提示驱动性通路活化在这些肿瘤的癌变过程中并不是必需的。但是由于缺乏可控的培养系统或动物模型,驱动性通路突变在人类CRC癌变中的具体作用非常难以评价。2011年,Sato等[4]开发了一种人类上皮细胞类器官体培养系统,利用人小肠干细胞(intestinal stem cells,ISCs)自我更新的能力,通过添加模拟干细胞龛微环境的各种因子,使ISCs在Matrigel中形成肠隐窝样类器官结构。2015年,他们进一步使用CRISPR-Cas 9基因组编辑技术,在正常人ISCs中依次突变掉APC、SMAD4、TP53、KRAS、PI3KCA基因,模拟了人类CRC中驱动性通路的活化状态,发现这种工程化类器官体的生长不再完全依赖于干细胞龛微环境因子,表明仅仅突变掉5个驱动基因的ISCs完全可以起始肿瘤的发生。但是将这种类器官体注射到小鼠脾脏,却仅能在肝脏形成微转移灶(<1 mm2)而无法形成肉眼转移灶,表明这种细胞的侵袭、转移能力有缺陷。进一步分析发现,这种类器官体细胞不具有其他的遗传异常,比如染色体异倍性、拷贝数改变或CpG岛甲基化表型等,其基因表达特征也跟人体CRC不完全一样,病理表现为高分化形态。然而,当他们在腺瘤分离的ISCs细胞中,同样突变掉这5个驱动基因后,经脾内注射即可形成大的肝内转移灶,表明除了这5个驱动基因突变外,人体CRC还具有其他的遗传特征[5]。那么腺瘤细胞跟正常结肠上皮细胞究竟哪里不同呢?最主要的区别是腺瘤细胞具有明显的染色体异常,即染色体不稳定性(chromosome instability,CIN)。
另外,临床上有一类特殊类型的腺瘤——无蒂锯齿状腺瘤(sessile serrated adenomas),这种腺瘤占结肠腺瘤的6~12%,好发于右半结肠,表现为宽的腔内锯齿状生长、结肠隐窝基部的分支和扭曲等特点,其癌变过程不完全遵循经典的“腺瘤——癌序贯模型”,反而表现为高频的BRAF、尾型同源盒基因2(caudal type homeobox 2,CDX2)突变和表观遗传学异常[6-8]。这一癌变过程也被称为锯齿状通路(serrated pathway),目前被认为是“腺瘤——癌序贯模型”的重要补充,约占全部CRC病例的15%左右[9]。因此,我们有必要全面地审视一下人体CRC细胞的遗传和表观遗传学特征。
一、人体结直肠癌细胞的遗传和表观遗传学特征
1. 高突变肿瘤的遗传特征——微卫星不稳定 性(microsatellite instability,MSI): 微 卫 星(microsatellite)是指基因组中广泛存在的1~4 bp的串联重复序列(重复次数通常在5~50次),人类基因组中平均每30 kb就有一个,其在DNA复制过程中的突变率明显高于其他DNA序列,因此在人群中具有高度的可变性和多态性,广泛的用于人群遗传学研究[10]。1992年,Perucho等[11]为了发现新的抑癌基因,用微卫星PCR分析了CRC和癌旁正常组织,发现一部分CRC肿瘤组织中的条带变短,位置下移了,进一步分析发现,这是由重复序列的碱基缺失造成的。1993年,这种现象被命名为 MSI[12]。
遗传性非息肉结直肠癌(hereditary nonpolyposis colorectal cancer,HNPCC),也称为Lynch综合征,也具有典型的MSI特征。其表现为早发性(通常在20~30岁)的结直肠、子宫内膜、胃、卵巢等的多发肿瘤。1993年Lynch综合征家系的遗传连锁分析,将致病基因定位到2p16的MSH2基因上[13-14],从而将MSI与DNA错配修复系统(DNA mismatch repair,MMR)联系了起来。
近年来的高通量测序研究发现,大约85%的CRC中,平均每个肿瘤有60个突变,而其余15%的CRC中,平均每个肿瘤有高达700个以上的突变,这部分肿瘤又被称作高突变肿瘤[7,15]。进一步根据突变产生机理的不同,将高突变肿瘤又分成两个亚类[16],第一个亚类约占高突变肿瘤的80%,突变率为15~40位点/Mb DNA序列,主要是由MMR系统缺陷造成的[17];第二个亚类约占高突变肿瘤的20%,其突变率在40位点/Mb DNA序列以上,又被称为超高突变肿瘤,主要是由DNA聚合酶ε(POLE)的外切酶活性结构域失活突变,导致校对功能缺失引起的,个别肿瘤还可由POLD1的校对功能缺陷引起[18]。
人类MMR系统包括9个基因——MSH2、MSH6、MSH5、MSH4、MSH3、MLH1、MLH3、PMS1 和PMS2。其中MSH2-MSH6、MSH2-MSH3组成的异源二聚体沿着新合成DNA链移动,识别DNA复制过程中的单碱基错配、小的插入/缺失错配环(insertion-deletion loop),并招募MLH1-MLH3、MLH1-PMS2、MLH1-PMS1等复合物切除错配并修复[19]。因此这一系统是生物体内DNA复制后的一种重要修复机制,在保证DNA复制的忠实性、维持遗传的完整性和稳定性方面具有重要作用。
Lynch综合征是由MMR基因的种系失活突变引起的遗传性疾病,目前公认的基因有4个,MSH2、MLH1、MSH6和 PMS2[20]。而大约 12%的散发型CRC有MMR缺陷引起的MSI,主要是由MLH1和PMS2蛋白的表达缺失引起。这类肿瘤中还常见MLH1基因启动子的双等位基因甲基化、高频的BRAF基因突变(通常在V600E位点),74%的肿瘤表现为二倍体。肿瘤还具有较为鲜明的临床病理特征,好发于右半结肠,表现为低分化、粘液型或印戒样形态,可见大量的T细胞浸润,患者预后好于其他非MSI的患者。
2. 染色体不稳定性(chromosome instability,CIN):CIN是指由于有丝分裂过程中染色体的错误分离引起高频的整条或部分染色体获得或缺失,由此导致不同子代细胞之间的核型异常[21]。最主要的表现是染色体数量的异常(异倍体)、部分染色体的扩增和高频的杂合性缺失(loss of heterozygosity,LOH)。造成CIN的原因目前还不完全清楚,但已知与染色体分离缺陷、端粒失功能、DNA损伤反应相关基因异常有关[22-23]。
CIN可见于65%~70%的散发性CRC,也是与“腺瘤——癌序贯模型”契合度最高的。CIN相关的CRC中常见多种癌基因和抑癌基因的累积突变,驱动基因APC、KRAS、SMAD4、PIK3CA和TP53在CIN相关CRC中的突变率分别为30%~70%、30%~50%、10%~20%、20% 和40%~50%[22]。那么CIN到底是细胞癌变的原因还是结果呢?目前认为,CIN在“腺瘤——癌序贯”通路的很早阶段就已发生,60%~80%的结肠息肉存在异倍体改变[24]。而APC基因的突变在CIN的形成过程中可能发挥始动作用,因为其除了在Wnt/β-catenin信号通路中发挥中心作用外,通过结合胞浆、纺锤体、中心体微管的正端,在细胞骨架调控中也发挥重要作用[22]。
具有CIN特征的CRC患者,无论种族、肿瘤的解剖部位、是否接受化疗等,比起具有MSI特征的患者,都具有较差的总生存和无进展生存率[22]。但是,由于具有典型的“腺瘤——癌序贯”癌变通路,因此为CRC的化学预防提供了机会。比如使用选择性的COX-2抑制剂可减少息肉的复发并降低CRC发生率,但却增加心血管病和肾性高血压的风险[25-27]。
3. CpG岛甲基化表型(CpG island methylator phenotype,CIMP):CIMP这一概念最早是由Toyata等[28]于1999年提出的,是指全基因组范围内,大量基因启动子区CpG岛自发性高甲基化的现象,导致多个抑癌基因或其他肿瘤相关基因的失活,反映了一种表观遗传学不稳定的状态。超过50%的人类基因表达可以通过启动子区CpG岛的甲基化状态来调节。与CRC有关的甲基化有两种:A型甲基化(又称年龄相关的甲基化)和C型甲基化(又称肿瘤特异的甲基化)[29]。A型甲基化与正常结直肠上皮细胞的衰老有关,受累基因多调控细胞的生长和/或分化,导致细胞癌变的易感状态。C型甲基化仅仅出现在肿瘤中,表现为CIMP。
CIMP相关的CRC同样具有鲜明的特征,30%~40%的右半结肠肿瘤和5%~15%的左半结肠和直肠肿瘤表现为CIMP,常见于女性、高龄、吸烟患者,病理以粘液型和低分化为主,而且肿瘤具有高频的BRAF基因突变[29-31]。但是CIMP对于患者预后的影响目前还存在争议,目前认为在去除其他临床因素和相关突变的影响后,CIMP与CRC患者的预后没有明显相关性[29,32]。
4. CRC肿瘤中可能同时共存多种遗传或表观遗传学特征:上述三个CRC癌变通路并不是完全互斥的,某些肿瘤中可同时存在多种特征,比如虽然大部分微卫星稳定的肿瘤是通过CIN通路癌变的,但大约25%的MSI相关CRC同时存在CIN表型,12%的CIN阳性的肿瘤同时存在高水平的MSI[33-34]。另外,CIMP常见于MSI阳性/CIN阴性的肿瘤,但大约33%的CIMP相关的CRC同时存在高水平的CIN[35]。
二、CRC的基因分型
由于不同癌变通路的CRC具有不同的临床病理特征,甚至是不同的预后和治疗反应性,因此随着个体化医学(personalized medicine)的发展,人们对CRC精准分子分型的需求越来越迫切。从2007年开始,科学家就尝试进行CRC的分子分型[36]。随着基因组学、转录组学及高通量测序等技术的发展,从2012年起,基于不同基因表达谱分析平台和分析方法的多个CRC分型系统相继被报道[3,37-43]。不同的分型系统之间有一定的相似性,比如MSI相关的肿瘤和间质基因高表达的肿瘤都被单独的分了出来,但是同样也存在很大的差异,不便于使用。为了规范CRC的基因分子分型,2015年,国际结直肠癌分型协作组(The CRC Subtyping Consortium,CRCSC)综合了六套CRC分型数据[37,39-43],开发了一套基于网络生物学的整合性分型算法,建立了4种CRC分子特征共识分型(consensus molecular subtype,CMS)——CMS1、CMS2、CMS3和 CMS4(表 1)[44]。四种分型在参与分析的近4 000例肿瘤中所占的比例分别是14%、37%、13%和23%,其余的是无共有特征性肿瘤,不具有任何一致性的特征,占到了全部原发性肿瘤的22%。
CMS1型又被称为MSI免疫型,表现为高的MSI和CIMP,低的CIN和强免疫原性。近70%的BRAF突变的患者都集中在这一型中。虽然高MSI的典型特征是高的基因突变率,但是除BRAF之外的其它常见CRC驱动基因,比如APC、TP53、KRAS、PIK3CA等的突变频率并不比其他类型高。这一类型肿瘤中常见的活化通路有JAK-STAT通路、Caspases通路等。肿瘤中常见免疫细胞的弥漫性浸润,主要为Th1细胞、细胞毒T细胞和NK细胞等;而且CTLA4、PD1、PDL1等免疫检测点分子高表达,具有高免疫原性,因此总生存和无进展生存较好,也是免疫检测点药物治疗的适宜人群。但此类患者一旦复发,其预后很差[44-45]。
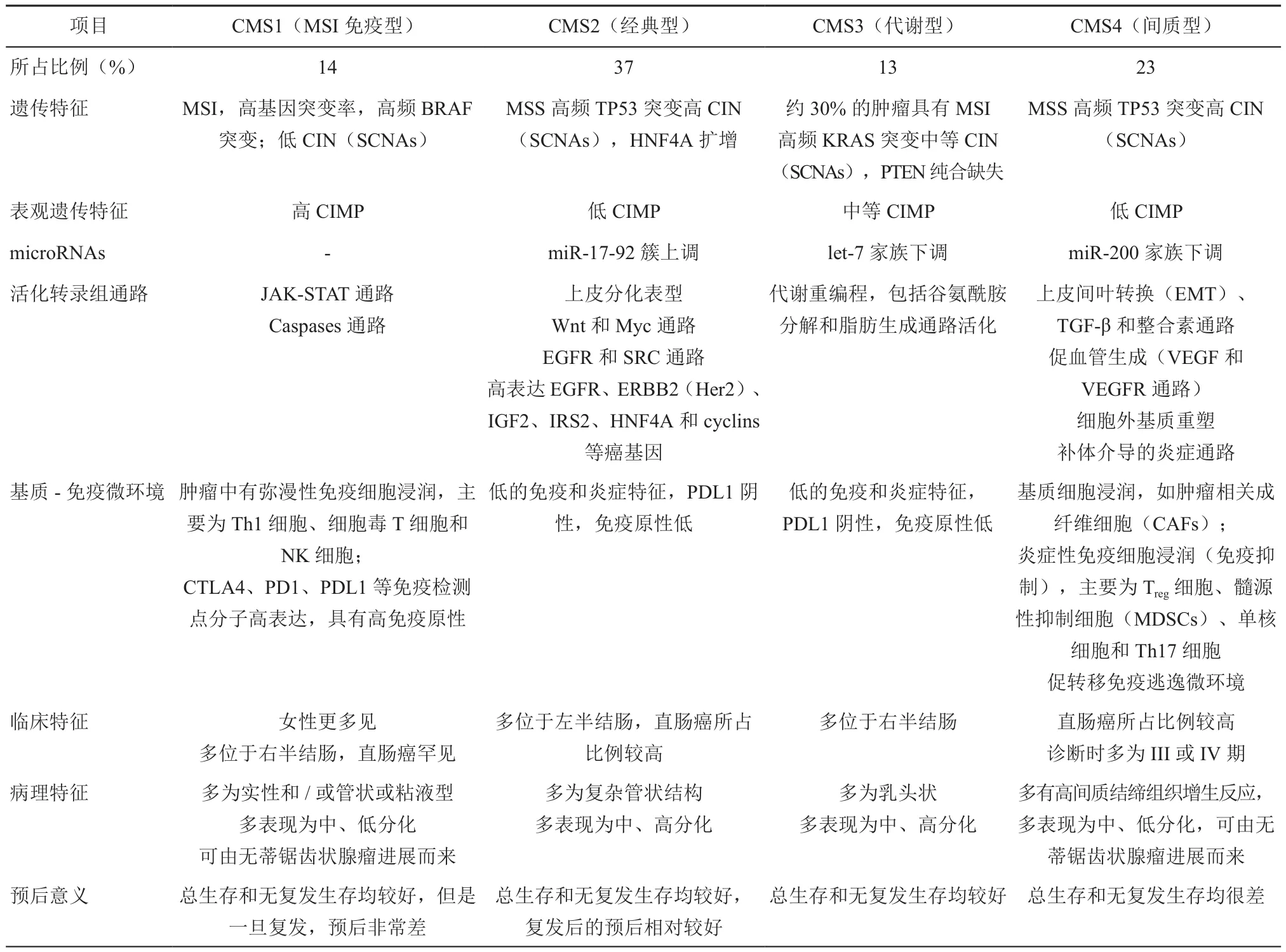
表1 CRC分子特征共识分型(CMS)的生物学和临床病理特征
CMS2型又被称为经典型,表现为高CIN、低CIMP和MSI特征,因此肿瘤具有典型的上皮分化特征,并存在大量的体细胞拷贝数改变(somatic copy number alterations,SCNAs)。比较特征性的是这一类型中15%的患者都具有HNF4A基因的扩增,而且Wnt和Myc靶基因,以及很多癌基因如EGFR、ERBB2 (Her2)、IGF2、IRS2、HNF4A和cyclins等过表达。常见的活化通路有EGFR和SRC通路、Wnt和Myc通路等。另外,此类肿瘤的免疫原性较低,但患者整体预后较好,即使是在肿瘤复发以后,其预后也优于其他类型的肿瘤[44-45]。
CMS3型又被称为代谢型,表现为中等程度的CIN和CIMP,30%的个体还具有MSI特征,KRAS突变的患者也在此型中相对富集。此型患者最突出的特征是细胞代谢谱的改变和代谢重编程,各种糖、脂、氨基酸、核苷酸代谢均处于活跃状态,尤其是谷氨酰胺分解和脂肪生成通路异常活化。CMS3型肿瘤的免疫原性同样较低,但患者整体预后较好[44-45]。
CMS4型又被称为间质型,表现为高CIN、低MSI和CIMP,它与CMS2型的主要区别是癌旁组织中有大量的基质细胞,如肿瘤相关成纤维细 胞(cancer-associated fibroblasts,CAFs); 肿瘤组织中也有大量的炎症细胞浸润,如Treg细胞、髓源性抑制细胞(myeloid-derived suppressor cells,MDSCs)、单核细胞和Th17细胞等。由于CAFs分泌高水平的TGF-β,而炎症细胞产生大量的炎症和免疫抑制因子,如CXCL12、CCL2、IL-23和IL-17等,因此表现出促转移的免疫逃逸微环境。另外,TGF-β信号通路以及上皮间叶转换(epithelial-mesenchymal transition,EMT)、整合素通路、VEGF和VEGFR通路、细胞外基质重塑和补体介导的炎症通路等均在此型中处于活化状态,因此患者的预后很差[44-45]。
此外,这四种分型的CRC也具有较为鲜明的临床病理特征(表1)[44-45]。CMS1型更常见于女性患者,肿瘤多位于右半结肠,直肠癌罕见,病理上多表现为中、低分化的实性和/或管状或粘液型;CMS2型多位于左半结肠,直肠癌所占比例较高,病理上多表现为中、高分化的复杂管状结构;CMS3型多位于右半结肠,病理上多表现为中、高分化的乳头状肿瘤;CMS4型中直肠癌所占比例较高,患者诊断时多为晚期,病理上多表现为中、低分化,并伴有高间质结缔组织增生反应。值得一提的是,绝大部分的直肠癌均为CMS2和CMS4型。
另外,通过对比管状腺瘤和无蒂锯齿状腺瘤的基因表达谱发现,无蒂锯齿状腺瘤中,TGF-β通路活化,依TGF-β通路活化程度的不同,可分别进展为CMS1和CMS4型肿瘤。通过BRAF激活突变建立的无蒂锯齿状腺瘤类器官体模型中,结合微环境中的高水平TGF-β信号,可直接驱动间质型CMS4样肿瘤的形成[46]。
三、CRC的蛋白质组分型
蛋白质是生命功能的最终执行者,也是联系基因型和表型的纽带。肿瘤相关的基因组和表观遗传学改变能否最终体现在蛋白质层面上,并指导患者分子分型?2014年临床肿瘤蛋白质组分析联盟(Clinical Proteomic Tumor Analysis Consortium,CPTAC)首先在CRC上对实体瘤的蛋白质组分型做了尝试[47]。他们对95对TCGA肿瘤组织样本进行了深度蛋白质组分析,平均每个样本鉴定到7 211个蛋白。与TCGA基因组和转录组测序数据的对比发现,比起种系DNA突变来,肿瘤组织内的体细胞突变更倾向于减少蛋白丰度,可能是因为降低了基因的翻译效率和蛋白稳定性。而拷贝数变异通常强烈影响mRNA的丰度,对蛋白丰度的影响则较弱。因此,mRNA和蛋白丰度的相关性较低,平均的Spearman相关系数只有0.23。这提示基因组和表观基因组的异常不一定能反映到蛋白层面上。
根据这些CRC患者肿瘤组织的蛋白质组表达特征,可以分成5个亚型——蛋白质组亚型A~E(表2)[47]。与基因组和表观基因组特征的比较发现,几乎所有高突变肿瘤都集中在亚型B和C中,亚型B中罕见TP53的突变和18q缺失,但明显与高CIMP表型相关,而亚型C中明显富集非CIMP表型个体和干细胞样特征。其余的三个亚型A、D和E,则与CIN特征相关,亚型E中明显富集TP53突变和18q缺失的个体,还与HNF4A扩增和高表达相关。
临床病理特征相关性分析显示,亚型C中明显富集II期肿瘤。标志蛋白的基因功能分析发现,亚型C的上调蛋白中明显富集于创伤反应、蛋白激活级联EMT相关的蛋白;亚型E的上调蛋白中明显富集于RNA代谢加工通路中。因此,亚型C与患者的不良预后相关。
虽然蛋白质组分型与CMS基因分型并不一致,但两者之间具有关联性[44]。比如,蛋白质组亚型A的个体主要富集在CMS2基因分型中;亚型B主要富集在CMS1型中;亚型C主要富集在CMS4型中;亚型D主要富集于CMS3亚型中;而亚型E主要富集在CMS2中。

表2 CRC蛋白质组分型的生物学和临床病理特征
四、CRC分子分型在精准医学诊疗中的意义
晚期CRC的靶向治疗开始于2004年,当时人们并没有考虑CRC本身的遗传特征,仅将肿瘤的共有特征——新生血管形成(angiogenesis)的关键基因VEGF作为治疗靶点[48]。随着对CRC分子癌变通路和遗传特征的认识,2009年起,标准化疗方案联合抗EGFR单抗(西妥昔单抗或帕尼单抗)的三期临床试验显示,KRAS基因野生型的患者对抗EGFR单抗的靶向治疗敏感,而KRAS第2外显子突变的个体对抗EGFR单抗原发耐药[49-50]。针对KRAS基因突变的分层模型开启了CRC精准治疗的时代,这一阶段也称为“单基因——单药物”模式[45]。
然而,多数KRAS基因野生型患者对西妥昔单抗和帕尼单抗同样不敏感,表明还存在其他的耐药机制。而且,其他单一靶向药物(如针对BRAF突变个体的BRAF抑制剂和针对KRAS突变个体的MEK抑制剂)的临床试验相继失败,提示针对单一位点的靶向治疗还存在一些弊端[51-52]。
高通量测序技术的发展推动CRC的精准靶向治疗进入“多基因——多药物”模式[45]。人们发现对EGFR抑制剂原发性耐药的肿瘤还受其他通路的影响,比如BRAF、MEK1、ERBB2、FGFR1和PDGFRA等[53]。而KRAS、NRAS、BRAF和PIK3CA均为野生型的肿瘤(也称四阴性肿瘤,约占CRC患者的30%),对EGFR抑制剂的治疗反应性更好[54]。四阴性肿瘤对靶向EGFR的酪氨酸激酶抑制剂(tyrosine kinase inhibitor,TKI)联合抗EGFR单抗的双重治疗特别敏感,这两种药物通过不同机制抑制EGFR,对于EGFR胞外结构域突变的肿瘤,双重EGFR靶向治疗同样有效[55-56]。另外,在四阴性肿瘤的患者源性异种移植模型(patient-derived xenograft,PDX)中,同时抑制EGFR和MEK能明显减少获得性耐药[57]。对于RAS野生型的肿瘤,尽早给予EGFR和MEK抑制剂治疗,能显著增加这条信号通路完全阻断的机会,减少获得性耐药。
对CRC分型认识的不断深入,使人们意识到不同CMS亚型具有显著的生物学差异,因此也会导致不同的药物反应性。CMS2型肿瘤因为具有EGFR配体和IRS2的高频扩增和/或过表达,抗EGFR靶向治疗通常可延长患者生存时间,而CMS1和CMS3型肿瘤的EGFR通路通常处于低活性或抑制状态,对抗EGFR靶向治疗无效[44,58]。另外,CMS2型肿瘤中还存在ERBB2和IGF2基因的扩增,因此联合使用ERBB和IGF1R的抑制剂可能会有更好的效果[59]。CMS1型肿瘤因为有强的免疫原性,高表达CTLA4、PD1、PDL1等免疫检测点分子,可能是PD1抗体治疗的适宜人群[44]。此外,CMS4型肿瘤中虽然RAS基因多为野生型,但是由于细胞呈现间叶细胞表型,因此对抗EGFR治疗产生治疗原发性耐药[44]。但联用西妥昔单抗和抗integrin-αv的单抗将使高表达integrin-αvβ6的CMS4型肿瘤患者获益[60]。针对CMS3型肿瘤,靶向代谢酶,如谷氨酰胺酶和脂肪酸合酶的新型抑制剂,正在研发阶段[61-62]。但是,针对不同CRC分型的分子基础,联合使用不同的靶向治疗策略,即“多分子——多药物”模式,正是未来CRC精准医学的核心[45]。
CRC的发生发展是一个漫长的生物学过程,涉及基因组、表观遗传组、转录组、蛋白质组,乃至代谢组等各个层面的大量基因、蛋白和小分子代谢物及其交互网络的异常改变。但是,现有的CRC分子分型系统还有赖于驱动基因突变检测、转录组和蛋白质组分析,受技术方法、数据分析和费用等因素的限制还难以大范围地应用于临床。相信随着对CRC分子癌变基础认识的不断深入,以及分型算法、工具的不断简化,CRC的精准分子分型必将在其精准诊治中发挥更加重要的作用。
猜你喜欢
中国临床医学影像杂志(2022年2期)2022-05-25
世界科学技术-中医药现代化(2022年2期)2022-05-25
皮肤病与性病(2021年3期)2021-07-30
癌变·畸变·突变(2021年2期)2021-04-15
天津医科大学学报(2021年2期)2021-03-29
祝您健康·文摘版(2020年11期)2020-11-11
癌变·畸变·突变(2020年5期)2020-10-19
中国动物检疫(2020年1期)2020-01-08
中国临床医学影像杂志(2019年6期)2019-08-27
