
PTB/nPTB:master regulators of neuronal fate in mammals
2018-10-17 06:18JingHuHaoQianYuanchaoXueXiangDongFu
Biophysics Reports 2018年4期
Jing Hu,Hao Qian,Yuanchao Xue,Xiang-Dong Fu,
1 Department of Cellular and Molecular Medicine,University of California,La Jolla,San Diego,CA 92093-0651,USA
2 Key Laboratory of RNA Biology,Institute of Biophysics,Chinese Academy of Sciences,Beijing 100101,China
Abstract PTB was initially discovered as a polypyrimidine tract-binding protein(hence the name),which corresponds to a specific RNA-binding protein associated with heterogeneous ribonucleoprotein particle(hnRNP I).The PTB family consists of three members in mammalian genomes,with PTB P1(PTB)expressed in most cell types,PTBP2(also known as nPTB or br PTB )exclusively found in the nervous system,and PTB P3(also known as ROD1)predominately detected in immune cells.During neural development,PTB is down-regulated,which induces nPTB,and the expression of both PTB and nPTB becomes diminished when neurons mature.This programed switch,which largely takes place at the splicing level,is critical for the development of the nervous system,with PTB playing a central role in neuronal induction and nPTB guarding neuronal maturation.Remarkably,sequential knockdown of PTB and nPTB has been found to be necessary and sufficient to convert non-neuronal cells to the neuronal lineage.These findings,coupled with exquisite understanding of the molecular circuits regulated by these RNA-binding proteins,establish a critical foundation for their future applications in regenerative medicine.
Keywords Polypyrimidine tract-binding proteins,Auto-and cross-regulation of alternative splicing,MicroRNA,Neuronal fate determination
INTRODUCTION
PTBwas originally identified as an RNA-binding protein with strong sequence-specific binding preference for the pyrimidine-rich tract located between the branchpoint sequence and invariant AGdinucleotide,which together constitutes a functional 3′splice site in pre-mRNA(Garcia-Blanco et al.1989;Patton et al.1991).PTB was soon recognized to correspond to hnRNPIin 2Dgel that could be immunoprecipitated as part of the heterogeneous ribonucleoprotein particle(Wang and Pederson 1990).Because PTB showed association with the spliceosome,and under certain conditions,was able to complement the splicing reaction,it was initially thought to function as an essential splicing factor(Ghetti et al.1992;Patton et al.1991).This proves not to be the case(Lander et al.2001),and for that matter,none of hnRNPproteins was later found to be essential for pre-mRNA splicing.However,PTB and nearly all hnRNP proteins have been shown to play roles in modulating splice site selection,thus functioning as splicing regulators in mammalian cells(Black 2003;Busch and Hertel 2012).
PTB has three paralogs in vertebrate genomes,sharing~70%sequence homology among them at the protein level,each containing four RNA recognition motifs(RRMs)(Fig.1).Biochemical and structural analysis showed that each RRM in PTB is able to independently bind RNA with similar preference for pyrimidine-rich motifs,suggesting the ability of PTB to create RNA looping(Oberstrass et al.2005).Additionally,RRM3 and RRM4 are able to interact with one another,which may be important for its function as a dimer or multimer in splicing control(Spellman andSmith 2006).Importantly,each PTB family member is expressed in a highly tissue-specific manner:PTBP1 is largely ubiquitously expressed in most tissues and cell types except neurons,whereas its paralog PTB P2 is restricted to neurons,and thus is also known as nPTB or brain-specific br PTB (Ashiya and Grabowski 1997;Lillevali et al.2001;Polydorides et al.2000).The third paralog PTB P3 has been identified as regulator of differentiation 1(ROD1)(Yamamoto et al.1999).As little is known about the function and action mechanism of PTB P3/ROD1,we focus here on PTB P1 and PTB P2,and for simplicity,we refer to them as PTB and nPTB,respectively,throughout this review.

Fig.1 Domains and comparison of three PTB family members in the human genome.Each PTB paralog contains four RNA recognition motifs(RRMs)responsible for protein-RNA and protein-protein interactions,the latter of which is thought to mediate PTB dimerization or multimerization during splicing control
FUNCTION OF PTB IN REGULATED SPLICING
The function of PTB in splicing control has been extensively studied on minigene models,which has been thoroughly reviewed earlier(Lander et al.2001;Spellman and Smith 2006).Given its preference for pyrimidine-rich motifs,it was initially thought that PTB acted as a splicing repressor by directly competing with U2AF65,a bona fide essential splicing factor that binds the polypyrimidine tract to initiate spliceosome assembly(Lin and Patton 1995;Singh et al.1995).However,additional analysis revealed that PTB often binds elsewhere in pre-mRNA(Lander et al.2001).Detailed mechanistic studies on the alternative c-src exon N1,which is normally excluded in non-neuronal cells,but included in neurons,demonstrated that PTB binds flanking intronic regions of N1 to interfere with the communication of this alternative exon with the downstream functional 3′splice site(Amir-Ahmady et al.2005;Chou et al.2000;Sharma et al.2005).On the Fas exon 6 minigene,however,PTB was found to bind within the alternative exon to prevent the recognition of functional splice sites on both sides of the alternative exon by the splicing machinery,which may result from PTB multimerization to create a silencing zone across the exon(Izquierdo et al.2005).
In the post-genome era,the function of PTB in splicing control has studied at the genome scale.Its binding profile was first determined in HeLa cells by crosslinking immunoprecipitation sequencing(CLIP)(Xue et al.2009).Interestingly,although PTB has been widely accepted asa splicing repress or,depletion of PTB in mammalian cells induced not only exon inclusion,as expected,but also exon skipping(Llorian et al.2010;Xue et al.2009).Integrated analysis of PTB binding and PTB -dependent splicing then revealed the position dependent effect of PTB ,which now applies to a large number of splicing regulators in mammalian cells(Corrionero and Valcarcel 2009;Fu and Ares 2014).In the case of PTB ,such ‘‘functional splicing map’’showed that PTB represses exon selection(thus its depletion causes exon inclusion)when it binds exonic and/or flanking intronic sequences around the alternative exon,whereas it promotes inclusion of many alternative exons(thus its depletion induces exon skipping)when it binds close to upstream constitutive 5′splice site and/or downstream constitutive 3′splice site,thereby strengthening the recognition of the alternative exon inbetween(Fig.2).
Relative to PTB ,much less is known about the role of nPTB in regulated splicing,and whether PTB P3/ROD1 plays a role in regulated splicing has remained an open question.In general,it is thought that nPTB functions similarly as PTB in splicing control,but as a weaker splicing repressor,in the nervous system because nPTB seems to be less effective than PTB in rescuing splicing defects of multiple splicing substrates examined in PTB and nPTB double-depleted cells(Boutz et al.2007b;Makeyev et al.2007;Spellman et al.2007).While the functional similarity between PTB and nPTB in splicing control is firm,as demonstrated by using PTB to replace nPTB in mature neurons in mice,these two PTB paralogs clearly have distinct functions in the development of the nervous system because PTB cannot fully rescue the phenotype caused by depletion of nPTB in developing neurons(Vuong et al.2016b).Such functional distinction requires further investigation,as PTB and nPTBmay enlist distinct co-factorsin splicing regulation or have entirely distinct functions beyond splicing control in different tissues or cell types.
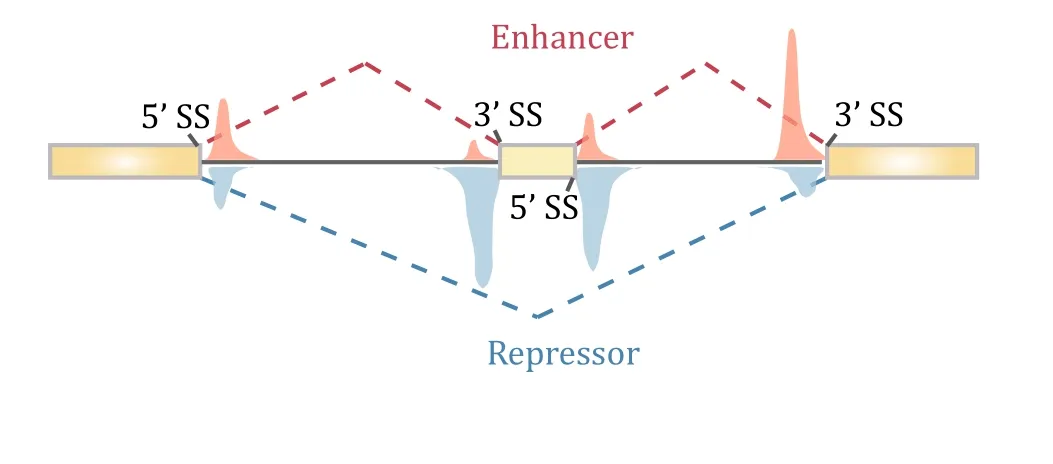
Fig.2 Position-dependent effects of PTB on regulated splicing.Compiling PTB -binding events on PTB -dependent exon inclusion events(red)versus PTB -dependent exon skipping events(blue)suggests that PTB action on flanking constitutive splice sites enhances the inclusion of the alternative exon in the middle,while PTB binding on or around the alternative exon causes skipping of the exon
AUTO-REGULATION AND CROSS-REGULATION OF PTB FAMILY MEMBERS
Depleting PTB or nPTB in cellular and animal models causes widespread changes in alternative splicing of a large number of genes(Coutinho-Mansfield et al.2007;Vuong et al.2016a).Because nPTB is exclusively expressed in the brain,its depletion has been found to affect a large array of neuronal-specific genes(Gueroussov et al.2015),including PSD-95 in mature neurons critical for excitatory synapse formation(Zheng et al.2012),and thus,such splicing defects are widely thought to contribute to specific neuronal phenotypes.Given so many splicing events are affected,however,it has been a major challenge in linking specific altered splicing events to defined phenotypic differences in most cases.Interestingly,sequential expression of PTB and nPTP during neuronal differentiation and characterization of their own regulated splicing programs have led to an elegant demonstration for auto-and crossregulation among PTB family members that are of direct functional relevance to the development of the nervous system(Boutz et al.2007b;Makeyev et al.2007;Spellman et al.2007).
It turns out that all three PTB family members themselves undergo alternative splicing in mammalian cells.In particular,PTB contains an alternative exon 11.PTB binds flanking intronic regions of this highly conserved alternative exon for its auto-regulation in non-neuronal cells(Fig.3A).This is because PTB represses the inclusion of exon 11 whose exclusion will alter the reading frame to create a premature termination codon(PTC)in exon 12,which will trigger nonsense-mediated mRNA decay(NMD)(Wollerton et al.2004).It has been estimated that NMD consumes~20%of PTB mRNA due to the suppression of its own exon 11.This ensures homeostatic expression of PTB in non-neuronal cells because increased PTB expression will reduce the inclusion of exon 11 to trigger NMD to reduce PTB expression,whereas decreased PTB expression will enhance the inclusion of exon 11 to produce more full-length mRNA to increase PTB expression.In developing neurons,however,PTB is progressively down-regulated due to the induction of the neuronal-specific microRNA miR-124(Boutz et al.2007b;Makeyev et al.2007).
Similar to PTB ,nPTB also carries an alternative exon 10,which is repressed by PTB in non-neuronal cells.As this exon is required to express a full-length functional nPTB,skipping of this alternative exon would prevent the expression of nPTB,as the resulting mRNA will be sensitive to NMD(Fig.3B).This affords the induction of nPTB when PTB expression is diminished in developing neurons,leading to progressive elevation of nPTB expression while PTB expression is gradually reduced.This cross-regulation demonstrates an elegant posttranscriptional strategy to switch the expression from PTB to nPTB during development of the nervous system,which proves to be functionally required(Coutinho-Mansfield et al.2007).
Last,but not least,PTBP3/ROD1 also carries an alternative exon 2,which is repressed by both PTB and nPTB,and the exclusion has been shown to introduce a stop codon in exon 3,thus may generate a potentially very short peptide(Fig.3C)(Spellman et al.2007).As PTB P3/ROD1 is mainly expressed in the immune system,it will be interesting to determine whether the development of the immune system also enlists a related strategy,as hinted by the name of ROD1.Together,such post-transcriptional strategy is responsible,in part,for tissue-specific expression of different PTB paralogs encoded in mammalian genomes.
ROLESOF PTB BEYOND SPLICING CONTROL
Besides its established functions in splicing control,PTB has been implicated in other processes of regulated gene expression,including polyadenylation(Castelo-Branco et al.2004;Moreira et al.1998),translation(Mitchell et al.2005),and mRNA stability(Hamiltonet al.2003;Pautz et al.2006).Relative to splicing control,however,our mechanistic understanding of such regulations has remained primitive.An intriguing possibility is that some of these splicing-independent functions of PTB may result from the role of PTB in antagonizing microRNA function,thereby modulating mRNA stability,as demonstrated during PTB -depletion induced neurogenesis(Xue et al.2013).
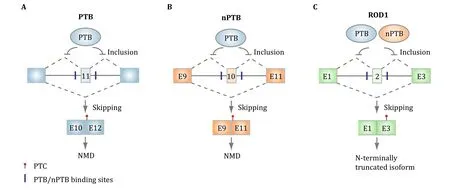
Fig.3 Auto-and cross-regulation of PTB family members at the splicing level.A PTB binds the flanking intronic sequences of the alternative exon 11 to auto-regulate its own expression,as exclusion of this exon will trigger NMD.B PTB also represses the inclusion of the alternative exon 10,which is required to generate full-length mRNA for nPTB in non-neuronal cells.PTBdown-regulation would thus induce this exon to produce functional nPTBduring neuronal development.C PTB and nPTB appear to function in a redundant fashion to repress the inclusion of the alternative exon 2 in PTB P3/ROD1,but the functional significance of this regulated splicing event remains to be determined
It has been well established that PTB is targeted by miR-124 during neuronal differentiation(Makeyev et al.2007),and nPTB by miR-133 during muscle differentiation(Boutz et al.2007a).Interestingly,based on the PTB CLIPdata generated on HeLa cells(Xue et al.2013),it is clear that PTB also finds pyrimidine-rich sequences in 3′UTR of various genes,some of which are coincident with specific microRNA responsive elements(Fig.4A).Indeed,by using reporter-based assays and through mapping Ago2 occupancy before and after PTB depletion in the genome,it became clear that PTB is able to directly compete with targeting of specific microRNAs on 3′UTR(Xue et al.2013).Therefore,in the absence of PTB ,those microRNAs become more efficient in inducing mRNA degradation and/or translational repression,which may also account for the observed effect of PTB in promoting insulin secretory granule biogenesis inβcells(Knoch et al.2004).Further interesting is the observation that PTB depletion not only destabilizes certain target mRNAs,but also exerts the opposite effect on others(Xue et al.2013).This turns out to be due to its role in modulating RNA secondary structure(Fig.4B).In certain cases,PTB binding disrupts such secondary structure to expose specific microRNA target sequences for their recognition by the microRNA machinery,and when PTB is depleted,such secondary structure is restored to prevent microRNA targeting,leading to mRNA stabilization.Together,these data suggest that PTB is both target and modulator of specific microRNAs in the cell,thereby building up additional layers in its regulated gene network during neuronal differentiation.It would be intriguing to investigate whether nPTB and ROD1 have similar microRNA modulation functions in future studies.
PTB/NPTB SWITCH DURING DEVELOPMENT OF THE NERVOUSSYSTEM

Fig.4 Modulation of microRNA targeting by PTB .A PTB binding directly competes with microRNA targeting on the microRNA responsive element(MRE).PTB depletion will thus promote microRNA targeting,thus enhancing microRNA-mediated posttranscriptional repression of gene expression.B PTB binding may also disrupt RNA secondary structure,which shields some MREs.PTB binding would expose such MREs for microRNA targeting,thereby enhancing post-transcriptional silencing of those genes
PTB is expressed in most cell types.Consistent with the ubiquitous expression of PTB and its broad function in regulated gene expression,genetic ablation of PTB resulted in embryonic lethality(Shibasaki et al.2013).Conditional knockout of PTB in the brain caused precocious neurogenesis of radial glial cells,a precursor cell type that gives rise to multiple neuronal cell lineages in the nervous system(Gotz and Barde 2005).This phenotype is actually related to the neuronal progenitor depletion phenotype caused by genetic inactivation of REST,a well-known master negative regulator of neurogenesis(Gao et al.2011;Singh et al.2008),thus in line with a key role of regulated PTB expression in development of the nervous system.Functional and mechanistic studies of PTB have been further pursued in embryonic stem cells and neuronal progenitor cells upon induction to the neuronal lineage,during which PTB expression is diminished and nPTB induced due to their cross-regulation(Boutz et al.2007b;Makeyev et al.2007).PTBis down-regulated by the induction of neuronal-specific microRNA miR-124,and such down-regulation has been found to be essential for neuronal induction,as demonstrated by the ability of a constitutively expressed exogenous PTB to block the process(Makeyev et al.2007).
Compared to the function of PTB in regulating neuronal induction,nPTB appears critical for neuronal maturation,as genetic ablation of this gene in the brain only compromised postnatal survival of knockout mice(Li et al.2014;Licatalosi et al.2012).Alarge number of adult neuron-specific alternative splicing events were affected in the developing brain,one of which characterized in detail corresponds to PSD-95,a key scaffold protein of excitatory synapses in mature neurons(Zheng et al.2012).Thus,the developmental switch from PTB to nPTB likely drives a programed switch of splicing control in development of the nervous system(Vuong et al.2016a).Morerecently,forced expression of PTB in nPTB null mice showed that PTB was able to rescue the phenotype of nPTB null mutation in forebrain,but not pan-neuronal knockout of nPTB in the brain(Vuong et al.2016b),implying that PTB can largely replace the function of nPTBonce neurons are fully matured,but not during the process of neuronal maturation.Although it has been postulated based on analysis of a set of PTB target genes that nPTB may act as a weaker splicing repressor than PTB (Makeyev et al.2007),the non-redundant function of nPTB in developing neurons raises the possibility that PTB and nPTB may have distinct targets,perhaps via different co-factors,as postulated(Vuong et al.2016b).It is also possible that nPTB may have functions beyond splicing control to drive neuronal maturation.
SUBTRACTING PTB AND NPTB TO TRANSDIFFERENTIATE NON-NEURONAL CELLS TO NEURONS
It has been demonstrated that certain key lineagespecific transcription factors(TFs)are able to drive the development of the nervous system,as various combinations of those TFs are able to not only induce neuronal progenitors to differentiate into functional neurons,but also convert non-neuronal cells to the neuronal lineage (Vierbuchen and Wernig 2012).Remarkably,diminishing PTB expression alone was found to be sufficient to initiate the entire neuronal differentiation program in diverse cell types of mouse origin(Xue et al.2013).Although diminished PTB expression is responsible for inducing a key neuronalspecific transcription factor Pbx1 through a splicing depression mechanism(Linares et al.2015),the vast majority of neuronal lineage-specific TFs appears to be induced through inactivating the REST complex(Fig.5A),thus qualifying PTB as a master regulator of the nervous system.
Mechanistically,non-neuronal cells are kept from becoming neurons by a negative feedback loop in which REST prevents the expression of a large number of neuronal-specific TFs as well as miR-124(Xue et al.2013).Interestingly,while miR-124 is able to target RESTin such loop,such targeting is potently suppressed by PTB in non-neuronal cells via direct competition of miR-124 targeting on multiple REST components,including a key REST subunit SCP1,a Pol II phosphatase(Xue et al.2013;Yeo et al.2005).Thus,the presence of PTB prevents the effect of precociously induced miR-124.Duringneuronal induction,increased miR-124,which likely results from signal-induced transcription in the brain,will target PTB to reduce its expression.Therefore,when PTB inactivation is experimentally induced by RNAi,such negative REST/miR-124 loop is converted to a positive one to allow miR-124 to be more efficient in targeting RESTand reduced REST further derepresses miR-124.In mouse cells,once this loop is activated,cells are progressively converted to functional neurons(Xue et al.2013).
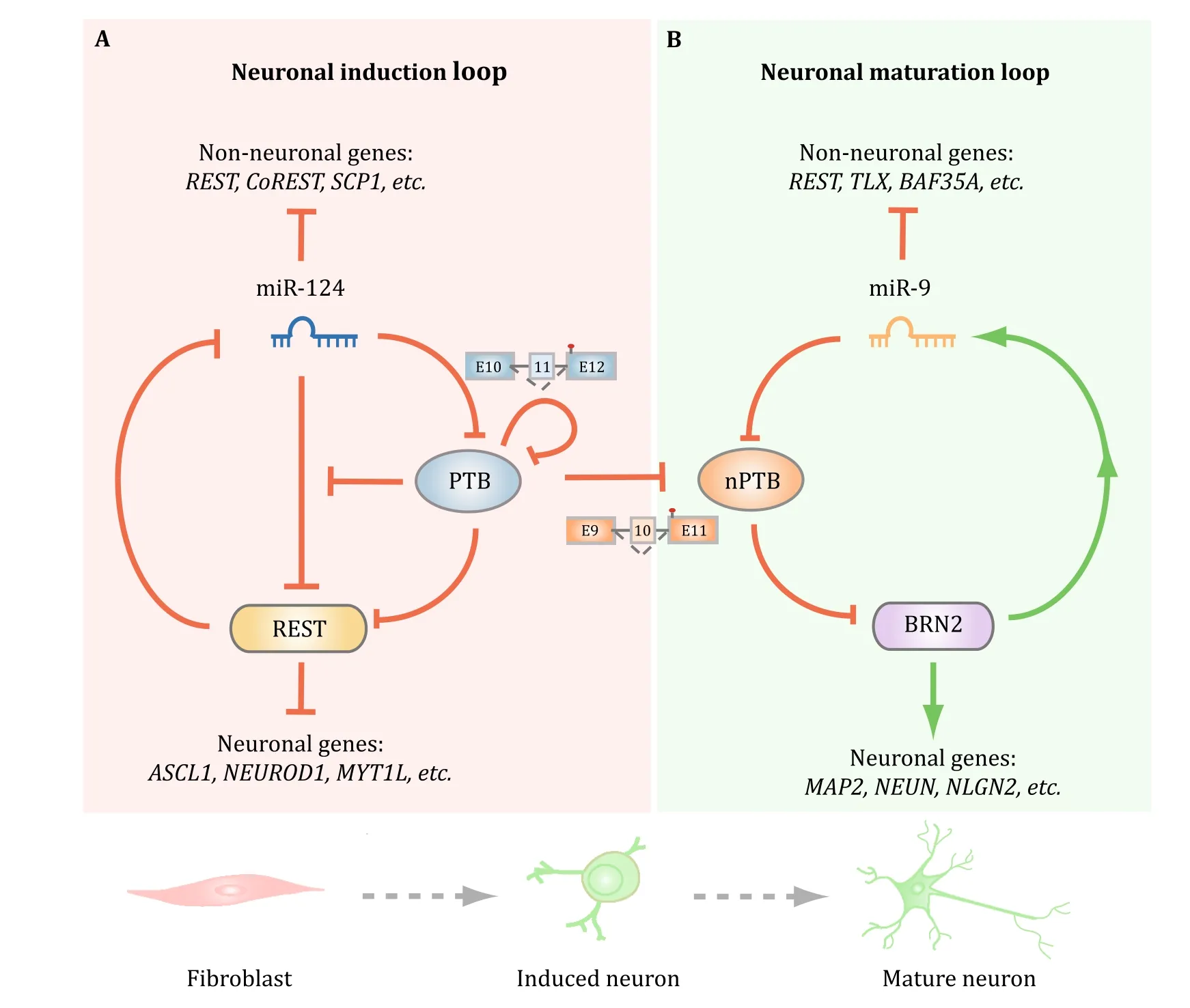
Fig.5 Regulation of two consecutive regulatory loops for neuronal induction and maturation by PTB and nPTB.A PTB functions as a breaker to miR-124 targeting of REST.Once PTB down-regulation is initiated,miR-124 becomes more efficient in targeting REST,and reduced REST further de-represses miR-124.This loop is self-enforced by two mechanisms because PTB itself is a target for miR-124 and reduced PTB also results in the inclusion of exon 11,together causing progressive PTB down-regulation during neuronal induction.B Reduced PTB expression de-represses nPTB due to the inclusion of the alternative exon 10.During neuronal maturation,induced miR-9 targets nPTB,which somehow leads to the induction of mature neuronal-specific transcription factor Brn2,and activated Brn2 further induces miR-9.Thus,PTB and nPTB function as two separate gatekeepers for neuronal induction and maturation.The two regulated loops are efficiently connected in mouse cells,likely because of high-level induction of miR-124,which is also capable of targeting nPTB,but the two loops have to be separately activated in human cells to generate functional neurons
When the same approach was applied to human adult fibroblasts,however,PTB inactivation by RNAi was found to be sufficient to propel neuronal induction,but insufficient to drive the process all the way to functional neurons(Xue et al.2016).This turns out to be due to persistent expression of nPTB in the absence of PTB in human cells,contrary to mouse cells in which nPTB is first induced and then diminished.Further analysis revealed another regulatory loop in which nPTB suppresses(directly or indirectly)the mature neuronspecific transcription factor Brn2,which activates miR-9,and this mature neuron-specific microRNA in turn targets nPTB(Fig.5B).Once this second loop is activated by sequential knockdown of PTB and nPTB or PTB knockdown in combination of Brn2 overexpression,all trans-differentiated neurons from human adult fibroblasts become fully matured in the presence of cocultured glial cells,showing all expected neuronal activities,including voltage-dependent Na+currents,repetitive action potentials,and both excitatory and inhibitory synaptic currents(Xue et al.2016).
Collectively,the experiments described above not only reveal the mechanism for nPTB down-regulation,but also suggest that the PTB -regulated REST/miR-124 loop is automatically connected to the second nPTB-regulated Brn2/miR-9 loop in mouse cells,but such two loops have to be separately activated in human adult fibroblasts.Such tight connection between the two loops in mouse,but not human cells might result from high-level activity of miR-124 in mouse,but not human cells because both miR-124 and miR-9 have been found to be capable of targeting nPTB(Xue et al.2016).The beauty of these regulatory networks by PTB and nPTB lies in the utilization of multiple regulatory mechanisms at both transcriptional and post-transcriptional levels,rather than simple feedback and/or feed forward controls among TFs.The PTB /nPTP-mediated neuronal reprogramming also emphasizes the ‘‘subtraction’’approach through inactivating negative master regulators compared to the general‘‘addition’’approach widely used in the regenerative medicine field via overexpressing positive master regulators.
ADVANTAGE OF MULTITASKING PTB IN CELL FATE DETERMINATION
Given that PTB is part of the regulatory loop to induce neuronal differentiation,such loop could be,at least in theory,induced by modulating any component in the loop,e.g.,inactivating REST or overexpressing miR-124,to achieve the same end purpose.As a matter of fact,inactivating REST or its key component SCP1 has been shown to induce neuronal-specific gene expression program(Yeo et al.2005).However,genetic ablation of REST has demonstrated that the complete loss of REST causes the depletion of neuronal progenitors and impairs viability of differentiated neurons(Gao et al.2011).Therefore,REST has to be maintained at an adequate lower level in differentiated neurons.As a result,the efficiency of siRNA against REST has to be titrated in trans-differentiation,as too low would be insufficient to achieve neuronal conversation,whereas too high would be detrimental to the viability of converted neurons.It is currently unclear how to experimentally reduce REST to an optimal level to induce neurogenesis.Thus,the strategy to down-regulate REST yet keep down-regulated RESTat the required low level is best achieved through the activation of the internal regulatory mechanism to maintain its hemostatic expression in differentiated neurons,such as the strategy of PTB knockdown.
Similarly,overexpressing miR-124 would lead to PTB down-regulation,thus activating the REST-miR-124 loop.However,it has been shown that miR-124 overexpression alone is insufficient to convert fibroblast to neurons(Yoo et al.2011).Even in neural stem cells(e.g.,N2a cells),overexpressed miR-124 is able to enhance neuronal differentiation induced by RA,but itself is not sufficient to drive the neuronal differentiation program(Makeyev et al.2007).This might be due to at least two technical reasons.First,in the published work on PTB knockdown-induced neurogenesis,PTB inactivation by siRNA is sufficient to trans-differentiate fibroblast into neurons,but the efficiency remains relative low,especially in human cells,which needs enhancement by a few small molecule inhibitors(Xue et al.2016).Additionally,as microRNAis not as efficient as siRNAin gene silencing in general,it is possible that transfected miR-124 alone is inefficient in down-regulating PTB to initiate the cell fate switch to the neuronal lineage.As optimized pools of small molecule inhibitors have been shown to be sufficient in inducing cell fate switch to different lineages(Ebrahimi 2016;Gao et al.2017),including from fibroblasts into neurons(Hu et al.2015;Li et al.2015),it is entirely possible that transfected miR-124 in combination with a cocktail of carefully titrated small molecular inhibitors might be able to drive neurogenesis.
For future therapeutic applications,especially in the brain,PTB/nPTB down-regulation appears to have multiple advantages over other strategies to induce neurogenesis.Because siRNA-or miRNA-based strategies suffer from challenging delivery problems,which has proven to be quite difficult in penetrating the brain blood barrier(BBB)(Mathupala 2009),the PTB inactivation-based strategy may bypass such technical problem by using ASO,which has been shown to effectively penetrate the BBB(Schoch and Miller 2017).Besides local delivery to minimize side effects,one can also envision additional ways to enhance the specificity by conjugating such anti-PTB ASO with a specific polypeptide to target desired cell types(Dowdy 2017).Thus,the PTB -based strategy may also be superior to small molecule-based approach for future applications in vivo,as the cell-type specificity of latter would be more difficult to manage.Because inactivation is clearly easier to achieve than overexpression,the ‘‘subtraction’’approach would be more feasible to implement than the‘‘addition’’approach using TFs in inducing transdifferentiation in vivo,despite the success in using various overexpressed TFs to directly convert nonneuronal cells,particularly astrocytes,to functional neurons in mouse brain(Grande et al.2013;Guo et al.2014;Liu et al.2015;Niu et al.2013;Su et al.2014;Torper et al.2013),which may offer considerable advantage over cell-based approaches in future therapeutic applications(Goldman 2016).As the development and differentiation of a neuron likely proceeds through its due course to orchestrate a series of switches during cellular reprogramming,PTB inactivation merely triggers the course to let the internal transcriptional and post-transcriptional programs to develop and progress.By contrast,forced expression of a TFor a combination of TFs may either be insufficient if not sufficiently overexpressed or detrimental to the internal program if excessively forced.
A PARALLEL OF PTB /NPTB-REGULATED CIRCUITRY IN THE MUSCLE SYSTEM?
PTB and nPTB have now been demonstrated to function as master regulators in the nervous system.Do these regulators also play roles in cell fate determination in other tissues?In fact,it has been found earlier that both PTB and nPTB are expressed in muscles and testis(Boutz et al.2007a).Parallel to the nervous system,PTB is highly expressed in myoblasts and its expression declines during muscle differentiation during which nPTB is induced,and interestingly,nPTB has also been found to subject to down-regulation by induced musclespecific miR-133,again parallel to its down-regulation by miR-9 in the nervous system(Xue et al.2016).A more recent single-cell transcriptome analysis also revealed a critical role of PTB as a key barrier to conversion from cardiac fibroblasts to cardiomyocytes(Liu et al.2017),although it is still under intensive debate in the cardiac field on whether cardiomyocytes are able to rise from cells of true non-cardiac linages,which has been argued against by using a rigorous double lineage tracing strategy(He et al.2017).It is also of interest to note that such a deterministic role of miR-133 in muscle differentiation has remained an open question,as an early study showed that this microRNA was able to enhance muscle differentiation in C2C12 cells(Chen et al.2006),but such effect was not observed in a separate study(Boutz et al.2007a),which suggests a fine-tuning role,rather than a deterministic function,of miR-133 in enhancing the function of differentiated muscles via a series of regulated alternative splicing events.This is reminiscent of a parallel enhancing,rather than deterministic function of miR-124 in neuronal stem cells.
REST is well known to dampen the expression of a large number of neuronal-specific genes in nonneuronal cells.It is interesting to note that NRF2 might fulfill such role as a master negative regulator in the muscle system,as it has been shown to repress some key muscle-specific genes,such as MyoD(Whitman et al.2013).As NRF2 has been well established to subject to a negative control by a dedicated ubiquitination system(Kansanen et al.2013),it is curious to investigate whether and how NRF2 down-regulation might be connected to diminished PTB expression during muscle differentiation.Similarly,we know little about nPTB with respect to its potential contribution to muscle differentiation.Considered together,it appears that there seems to be a parallel PTB /nPTB-regulated circuitry in the muscle system,which clearly requires further investigation,and when properly developed,such circuitry might be developed as tools to learn muscle fate determination and to engineer transdifferentiation from non-muscle cells to muscle or to replenish lost cardiomyocytes in failing heart.
Last,but not the least,given the success in elucidating PTB -and nPTB-regulated gene networks in cell fate determination in the nervous system,the question is whether other RNA-binding proteins or families of them play similar roles in other cellular reprogramming processes.In this regard,the toolbox of~1500 RNA-binding proteins expressed in mammalian cells(Gerstberger et al.2014)remains to be explored.
AcknowledgmentsThis work was supported by grants from the National Key R&D Program of China(2017YFA0504400)and National Institutes of Health of the US(GM049369 and HG004659).
Compliance with Ethical Standards
Conflict of interestJing Hu,Hao Qian,Yuanchao Xue,Xiang-Dong Fu declare that they have no conflict of interest.
Human and animal rights and informed consentThis article does not contain any studies with human or animal subjects performed by any of the authors.
Open AccessThis article is distributed under the terms of the Creative Commons Attribution 4.0 International License(http://creativecommons.org/licenses/by/4.0/),which permits unrestricted use,distribution,and reproduction in any medium,provided you give appropriate credit to the original author(s)and the source,provide a link to the Creative Commons license,and indicate if changes were made.

- Biophysics Reports的其它文章
- The road to a first-class research institute: creating an exceptional environment, recruiting outstanding scientists,producing high-impact work, and nurturing a culture of innovation-Preface to the 60th anniversary special issue
- Roles of H3K36-specific histone methyltransferases in transcription:antagonizing silencing and safeguarding transcription fidelity
- Cancer stem cells and tumorigenesis
- Structural roles of lipid molecules in the assembly of plant PSII -LHCII supercomplex
- Hessian single-molecule localization microscopy using sCMOS camera
- Dissection of structural dynamics of chromatin fibers by single-molecule magnetic tweezers