
Population Size, Genetic Diversity and Molecular Evidence of a Recent Population Bottleneck in Hynobius chinensis, an Endangered Salamander Species
2018-09-27 05:43EricGilbertKAZITSAShichaoWEIYunhaiPUXingyanWULinSONGLeiGAOFuyuanQIUYueGUOZhaoquanZHUandHuaWU
Asian Herpetological Research 2018年3期
Eric Gilbert KAZITSA, Shichao WEI, Yunhai PU, Xingyan WU, Lin SONG, Lei GAO, Fuyuan QIU, Yue GUO, Zhaoquan ZHU and Hua WU*
1 Institute of Evolution and Ecology, School of Life Sciences, Central China Normal University, 152 Luoyulu, Hongshan District, Wuhan 430079, China
2 Wildlife Conservation Station of Hubei Province, 438 Xiongchu Avenue, Hongshan District, Wuhan 430079, China
Abstract Severe population declines can reduce species to small populations, offering permissive conditions for deleterious processes. For example, following such events, species can become prone to inbreeding and genetic drift which can lead to a loss of genetic diversity and evolutionary potentials. Hynobius chinensis is a poorly studied very rare and declining endangered amphibian species endemic to China in Changyang County. We investigated adult census population size by monitoring breeding populations from 2015 to 2018, developed microsatellite markers from the transcriptome and used them to investigate genetic diversity, and a population bottleneck in this species.We found H. chinensis in 4 different localities in a total area of 2.18 km2 and estimated the overall adult census population size at 386-404 individuals. The adult census size (mean ± SE) per breeding pond ranged from 44 ± 6 to 141 ± 8 individuals and appeared smaller than that reported in closely related species in undisturbed habitats. We developed and characterized 13 microsatellite markers in total. Analysis of data at 7 loci (N = 118) in Hardy-Weinberg equilibrium gathered from the largest population showed that genetic diversity level was low. The average number of alleles per locus was 2.14. The observed and expected heterozygosities averaged 0.38 and 0.40, respectively. The inbreeding coefficient was -0.06. All tests performed to investigate a population bottleneck, i.e. The Garza-Williamson test, Heterozygosity excess test, Mode shift test of allele frequency, and effective population size estimates detected a population bottleneck. The contemporary and the historical effective population sizes were estimated at 36 and 234 individuals, respectively. We argue that as bottleneck effects, the studied population may have become prone to genetic drift and inbreeding, losing microsatellite alleles and heterozygosity. Our results suggest that populations of H. chinensis may have been extirpated in the study area.
Keywords transcriptome, microsatellites, adult census population size, effective population size, genetic drift,inbreeding
1. Introduction
Elucidating factors and processes affecting genetic diversity in species of conservation concern can help understand the ecology and evolution of such species.It can also help formulate appropriate conservation strategies (Amos and Balmford, 2001; Frankham, 2005).
Theoretical and empirical works show that severe population declines or population bottlenecks can affect genetic diversity in several ways (Amos and Balmford,2001; Frankham, 2005; Hoffmannet al., 2017; Willi and Hoffmann, 2009). For examples, by reducing species to small populations, population bottlenecks can reduce the standing genetic diversity and impair the ability to fix novel adaptive genetic mutations (Frankham,2005; Hoffmannet al., 2017). Still, these demographic events can offer permissive conditions for inbreeding and random drift, which can cause a continuous loss of genetic diversity and of individual fitness or inbreeding depression (Amos and Balmford; Frankham, 2005;Heredia-Bobadillaet al., 2017; Jordanet al., 2009; Spearet al., 2006; Storferet al., 2014; Willi and Hoffmann,2009). Due to these effects, affected species can lose their ability to respond to novel selection pressures and adapt novel environmental conditions (Frankham, 2005;Hoffmannet al., 2017; Willi and Hoffmann, 2009).Population bottlenecks can also render species vulnerable to extinction due to stochastic and catastrophic events(Amos and Balmford, 2001; Frankham, 2005; Willi and Hoffmann, 2009).
Severe population bottlenecks cause the values of different population genetics parameters, including heterozygosity and allele frequency at selectively neutral loci such as microsatellites to differ from those expected at Hardy-Weinberg equilibrium across several generations(Garza and Williamson, 2001; Luikartet al., 1998; Peeryet al., 2012). Different tests based on the analysis of multilocus microsatellite data within populations allow detecting severe population bottlenecks that occurred in the recent history of species by detecting violation of theoretical expectations under this equilibrium (Garza and Williamson, 2001; Luikartet al., 1998; Peeryet al.,2012). These include the Mode shift test, the Garza-Williamson test (the updated version of the GarzaM-ratio test), and tests of Heterozygosity excess (Garza and Williamson, 2001; Luikartet al., 1998; Peeryet al.,2012).
Potential factors causing population bottlenecks and loss of genetic diversity may vary between organisms (Rivera-Ortízet al., 2015). For amphibians,anthropogenic destruction and degradation of natural habitats is probably among the most important (Allentoft and O’Brien, 2010; Rivera-Ortízet al., 2015). Studies that analyze microsatellite data in amphibian species whose natural habitats have been considerably reduced and degraded by human activities show that such species have undergone fragmentation in gene pools. They have reduced census and effective sizes and exhibit molecular signs of recent population bottlenecks, inbreeding and loss of microsatellite alleles and heterozygosity (Allentoft and O’Brien, 2010; Heredia-Bobadillaet al., 2017;McCartney-Melstad and Shaffer, 2015; Rivera-Ortízet al.,2015; Storferet al., 2014; Sugawaraet al., 2015).
The Chinese salamander,Hynobius chinensis(Caudata,Hynobiidae) is an amphibian species endemic to China(Fei and Ye, 2016; Frost 2018). Classified as Endangered by the IUCN Red List (IUCN, 2018), literature shows thatH. chinensisis threatened by anthropogenic habitat degradation and destruction, causing habitat loss for populations (Fei and Ye, 2016). As effects of these threats,this species has undergone population declines and is very rare (Fei and Ye, 2016; IUCN, 2018). It is restricted to mountain forests in Changyang County in Yichang(Hubei province) -Note that Changyang County covers 3 412 km2(Source: Wikipedia)- at altitudes ranging from 1 400 to 1 500 m (Fei and Ye, 2016; Frost, 2018).
H. chinensisis a poorly known and studied species.Data on many aspects of Biology, including landscape use, genetic diversity, current population size, and demographic history are scarce.H. chinensishas been mostly involved in studies addressing systematic and taxonomic issues (Fei and Ye, 2016; Frost, 2018; Wanget al., 2007).
The nature of threats facingH. chinensis, the level of rarity, and the declining population trend reported in the literature let suspecting that this species might have experienced severe population bottlenecks and lost genetic diversity in the recent past (Apodacaet al.,2012; Heredia-Bobadillaet al., 2017; Sugawaraet al.,2016; Sugawaraet al., 2015; Storferet al., 2014; Wanget al., 2017). If the history ofH. chinensisconforms to this hypothesized trajectory, we should expect to see representative populations of this species exhibiting molecular signs of population bottlenecks detectable by microsatellite markers, low levels of genetic diversity and reduced sizes (Apodacaet al., 2012; Heredia-Bobadillaet al., 2017; Storferet al., 2014; Sugawaraet al., 2016;Sugawaraet al., 2015; Wanget al., 2017).
We investigated (i) adult census population size,developed microsatellite markers and used them to investigate (ii) genetic diversity, and (iii) molecular signatures of a population bottleneck inH. chinensis.Our aim was to contribute to our understanding of the demography and genetic viability of this endangered species.
2. Materials and Methods
2.1. Study area and field procedurePublished literature shows thatH. chinensishas one breeding period per year which begins in November (Fei and Ye, 2016). During the breeding period, adult individuals aggregate, mate and reproduce in ponds (Fei and Ye, 2016). An individual female lays eggs in a pair of sacs, with an egg sac’s length measuring tens of cm (Fei and Ye, 2016).
From November 2015 to April 2016, we searched forH. chinensisindividuals and egg sacs in ponds at altitudes ranging from 1300 to 1600 m in Changyang (Fei and Ye, 2016; Frost, 2018). Egg sacs were considered as a precursory sign of the presence ofH. chinensisas this species is the only Hynobiid salamander distributed in Changyang (Fei and Ye, 2016). We found breeding populations ofH. chinensisin at different sites of different localities.
Data fromH. nebulosusshowed that the number of adult male individuals could be accurately determined by examining breeding aggregations because a male individual would spend several weeks in a breeding pond(Kusano, 1980). We examined a breeding population ofH. chinensisat a breeding pond found in one locality:Dongtoutang. The pond was small and shallow (Table 1). And, the pond’s water was enough clear to observe objects present in the pond. Based on our previous field experience, and on information provided by local people, we were expecting to observe the largest breeding population at this pond. At the landscape scale, the area was at 50% covered by a degraded natural deciduous forest ofQuercussp. (Fagaceae). The pond’s surrounding area was covered by various herbs (mainly Poaceae,Oxalidaceae, and Polygonaceae). The minimum distance to a forest patch was 25 m.
H. chinensisindividuals were carefully collected from the pond, using a net (Heyeret al., 1994). The salamanders were sexed based on the morphology of the cloacae (Fei and Ye, 2016), marked by toe-clipping and released (Heyeret al., 1994). The number of captured individuals was recorded for each sex. After a time period ranging from of 1 to 4 weeks, the population was re-examined. For each sex, the number of “captured”,i.e. non-toe-clipped, and the number of “recaptured”i.e. toe-clipped individuals were recorded (Heyeret al.,1994). Pairs of egg sacs were progressively individually tagged as they were spawned and their total number was determined at the end of the breeding season (Chenet al.,2016; Kusano, 1980). The investigation was finished on March 25, 2016. There was no additional novel pair of egg sacs since March 2; and no unmarked male individual since March 8, 2016. Then, the number of adult females,Fand the number of adult males,Mwere estimated as the number of pairs of egg sacs (Chenet al., 2016; Kusano and Miyashita, 1984; Kusano, 1980) and toe-clipped male individuals, respectively (Heyeret al., 1994). The sex ratio,SRwas estimated asSR=M/Fand the adult census population size,NcasNc =M+F.
To minimize the impact on the populations(McCartney-Melstad and Shaffer, 2015), as the value ofSRwas close to that reported in closely related species(Kusano, 2012), for the breeding populations found in other localities and for the remaining study period, the values ofNcwere indirectly estimated. In summary, for each population, the number of adult females,Fnwas estimated based on egg clutch number as previously explained. And,Ncnwas estimated as:Ncn=Fn + (Fn*SR),wherenrepresents a given population. For all the localities, the values ofNcwas determined for 2015-2016;2016-2017 and 2017-2018 breeding seasons, at the same ponds and following this method.
From 22 different egg clutches, 22H. chinensishatched larvae samples were randomly collected. All these larvae and the toe clips were kept in anhydrous (98%)ethanol at -20 °C in the laboratory for DNA-based experiments.
During the fieldwork, to identify threats facingH.chinensisin the study area, local people were interviewed for a better understanding of the evolution of landscapes and of the usage of the species by humans. The species’mating behavior was occasionally observed at the breeding pond found in Dongtoutang.
2.2. Development of microsatellite markersAn adultH. chinensisindividual was brought to the laboratory,euthanized by injection of MS-22 (3g/l), and dissected.A tissue sample was taken from the liver. Total RNA was extracted (Omega Bio-Tek, USA) with TRIzol Regent®(Invitrogen, CA, USA). The purity, concentration and integrity of RNA were assessed by Nanodrop, Qubit 2.0 and Agilent 2100 bioanalyzer, respectively. Using 1μg of total RNA as input, a library for Illumina sequencing was prepared following the NEBNext Ultra™ RNA protocol.The library preparation was sequenced on an Illumina HiSeq 2500 platform (pair-end reads, 125 bp).
Sequencing results were analyzed and reads containing adaptors, reads containing poly-N, and low-quality reads(i.e. with more than 50%Q≤ 10 bases) were filtered out.And, the clean reads (i.e. the remaining reads) wereDe novoassembled, using Trinity v2.0.6 software (Grabherret al., 2011).
The Simple Sequence Repeat Identification Tool available at http://archive.gramene.org/db/markers/ssrtool/ was used to identify microsatellite loci in the assembledH. chinensis’ unigene DNA sequences.The following searching criteria were applied: (i) the minimum number of repeats: 5 times; (ii) the repeat type:tetranucleotide; and (iii) flanking sequence length: ≥ 18 bp. Using the software Primer v3.0, 74 microsatellite loci were screened and 119 primer pairs were designed,applying the following criteria: (1) primer length 18-23 bp, with 20 bp as optimum; (2) product size (bp): 100-250 bp; (3) primer melting temperature: 55-69 ℃ , with 60 ℃ as optimum and (4) GC% content: 40-70%, with 50% as optimum, and (5) GC clump: 2.
Genomic DNA was extracted from all the toe tissue and larvae samples, using TIANamp Genomic Kit(TIANGEN). The 119 primer pairs were synthesized(Applied Biosystems, Beijing, China) and tested by gradient PCR on the standard agarose gel. Individual PCR amplification was performed as following: an initial denaturation at 94 °C for 5 min was followed by 35 cycles of denaturation at 94 °C and at a primer’s specific annealing temperature for 30 sec, and an extension at 72°C for 45 sec and finally at 72 °C for 8 min. PCR was performed in a reaction volume of 10 μl containing 0.6 μl of template DNA, 5 μl of Taq DNA polymerase (Premix),of 0.3 μl of forward primer, 0.3 μl of reverse primer, and 3.8 μl of ddH2O. In total, 51 primer pairs consistently amplified at optimal annealing temperature and produced clear bands in the expected size range. These primer pairs were retained for polymorphism test.
The 5' end of each forward primer was labeled with one of the fluorescent dyesFAM,TAMRAorHEX, and polymorphism was tested, using data from the 22 larvae samples. In summary, PCR amplification of the 51 primer pairs was performed, following the protocol above described. For each individual larva, genotyping at the 51 microsatellite loci was performed with an ABI Prism 3730 Genetic Analyzer (Applied Biosystems, Beijing, China).Microsatellite alleles were scored, using GeneScan ROX 400 size standard and GeneMapper v4.0 software(Applied Biosystems, Beijing, China). The existence of genotyping errors (i.e. stuttering, large allele dropout) and of null alleles within the microsatellite data (N= 22) was checked, using the software Micro-Checker v2.2.3 (Van Oosterhoutet al., 2004).
An exact test for Hardy-Weinberg equilibrium(HWE) for each locus and a test for potential linkage disequilibrium (LD) between all pairs of loci were implemented in the online Genepop software (Raymond and Rousset, 1995), using Bonferroni-correctedP-values(Rice, 1989). ExactP-values were determined by Markov Chain method, using the following settings:dememorization: 10 000; batches: 100; iteration per batch:1 000 (Raymond and Rousset, 1995). The values of basic genetic diversity indices, exclusion probabilities, and polymorphism information content (PIC) were determined for each locus and for overall loci, using Cervus v3.0 software (Kalinowskiet al., 2007). Genotypes data were analyzed and the sampledH. chinensisadult individuals were genotyped at polymorphic loci in HWE, without LD,null alleles and/or genotyping errors (Table 2), following the procedure above described.
2.3. Analysis of genetic diversity and bottleneck effectsFor the adult samples’ genotype data, the observed heterozygosity (Ho), expected heterozygosity (He), and the number of alleles (A) were calculated for each locus and for the overall loci, using Arlequin v3.5 software(Excoffier and Lischer, 2010). The inbreeding coefficient,Fiswas calculated for each locus and for the overall loci,using FSTAT v2.9.3 software (Goudet, 2001).
Different approaches were used to investigate genetic signatures of a population bottleneck. First, the signature of a population bottleneck was tested by Garza-Williamson test. In summary, Garza-Williamson index,GWwas estimated for each locus asGW=k/(r+1), wherekis the number of alleles, andrthe allele size range(Excoffier and Lischer, 2010). And, the overallGWvalue was compared to the theoretical critical value for rejecting the null hypothesis of demographic stability that is 0.68 (Garza and Williamson, 2001; Peeryet al.,2012). The value ofGWis expected to be below this critical value in bottlenecked populations. The core idea behind Garza-Williamson test is that during population bottleneck, reduction inkis proportionally important than the reduction inr. Hence, a mall value ofGWcan be considered a genetic signature of a population bottleneck(Excoffier and Lischer, 2010; Garza and Williamson,2002). The Garza-Williamson statistics were computed in the software Arlequin v3.5 (Excoffier and Lischer, 2010).
Second, the signature of a population bottleneck was investigated based on heterozygosity excess (Cornuet and Luikartt, 1996), using Bottleneck v1.2.02 software(Luikart and Cornuet, 1999). In Bottleneck v1.2.02,three different mutation models, namely the infinite allele model (IAM), the stepwise mutational model(SMM), and the two-phase mutational model (TPM); and three different statistical tests namely the Sign test, the Standardized difference test and the Wilcoxon signedrank test are available for heterozygosity excess testing(Luikart and Cornuet, 1999). Considering the number of used loci, heterozygosity excess was tested by the Wilcoxon signed-rank test, under all mutation models.The following settings were applied: TPM with SMM accounting for 95% of mutations, 5% SMM, a variance among multiple steps of 12 000 and 10 000 iterations(Luikart and Cornuet, 1999).
Third, genetic signature of population bottleneck was investigated by Mode-shift test (Luikartet al.,1998), using the software Bottleneck v1.2.02 (Luikart and Cornuet, 1999). A putatively stable population is expected to have a peak of allele number at the lowest frequency class, resulting in an L-shaped distribution of allele frequency. Population bottleneck displaces the peak at other frequency classes, causing distortion of this graphical pattern or a shifted mode of allele frequency distribution (Luikartet al., 1998).
Finally, the trajectory of the effective population size on long-term was investigated by comparing the historical and the contemporary values. Assuming mutation-drift equilibrium, the historical effective population sizeNewas estimated based on heterozygosity data asNe = (1/[1-He]2-1)/8μ, whereHeis the average expected heterozygosity andμthe mutation rate per generation (Nei, 1987).Forμ, 10-3, the value reported in other amphibians was used (Peeryet al., 2012). The contemporaryNewas estimated based on LD, using the single-sample method as implemented in NeEstimator v2.0 software (Doet al.,2014). A potential bias from rare alleles was controlled and apcriticvalue of 0.01 was used (Doet al., 2014).LD estimates are based on the expectation that asNedecreases, genetic drift systematically associates alleles at different loci. Hence, in bottlenecked populations, the level of LD reflectsNe(Doet al., 2014).
2.4. Ethic statementAccess to the focal species and the fieldwork did not require any formal research permit.The animal manipulation and sampling protocols were previously approved by the Animal Welfare Committee of the Central China Normal University.
3. Results
3.1. Adult census population sizeWe foundH.chinensisat 4 sites of different localities (Figure 1; Table 1), in a total area of 2.18 km2. These sites were located north-east of Yichang city (Figure 1). The overall adult census population size in the area slightly varied between breeding seasons. It ranged from 386 to 404 individuals(Table 1). Reproduction was completed in permanent ponds of varying sizes (Table 1). These ponds occurred at altitudes ranging from 1 408 to 1 582 m (Table 1) and were distant from 1.01 to 3.2 km.
In Dongtoutang during the 2015-2016 breeding season, we identified 62H. chinensisadult females and 78 adult males, resulting in anNcof 140 individuals and a sex ratio of 1.26:1. We sampled 118 adult individuals in total, including 40 females and the 78 males.H. chinensisindividuals appeared in the breeding pond found in this locality on November 15. The number of male individuals found in the pond increased from the first (November 26,2015) to the third (January 4, 2016) census, reaching a peak of 60 individuals (Figure 2). The peak lasted till the sixth (February 20, 2016) census with minor fluctuation(Figure 2). The number of male individuals found in the pond then decreased till the last (March 8, 2016) census(Figure 2).
As expected, we obtained a high recapture rate of male individuals (Figure 2). Out of the 78 male individuals identified in total, 67 (85.90%) were already identified and toe clipped on January 4, 2016, i.e. with the third census (Figure 2). Since the fourth census on January 13, 2016, 97.30-100% of all male individuals found in the breeding pond were individuals that had already been identified and toe clipped (Figure 2). As expected,together, this high recapture rate and the evolution pattern of the number of male individuals found in the pond along the breeding season suggest that male individuals spent several days in the breeding pond. They confirmed the accuracy of the method used to estimate the number of adult males andNc.
The average value (± SE) ofNcover the study period(i.e. 2015-2018) varied between localities or breeding ponds. It ranged from 44 ± 6 to 141 ± 8 individuals (Table 1). And, as expected, the maximum value ofNcwas observed in Dongtoutang (Table 1). The sex ratio was 1.26: 1.
3.2. Microsatellite markersWe obtained 27 721 722(91.33%) clean reads in total. The Trinity assembly resulted in 77 111 unigenes. The size of the unigenes ranged from 201 to more than 3 000 bp (Figure 3). The total length of the unigenes and N50 were 46 797 297 bp and 951 bp, respectively. Among the unigenes, 46 378(61.14%) were not more than 400 bp in length. A total of 20 599 (26.71%) unigenes were in the range of 401-1000 bp. Unigenes longer than 3 000 bp were 1 507 (2.04%) in total (Figure 3).
The transcriptomic sequencing raw data have been deposited into the NCBI Sequence Read Archive (SRA)database under the accession number SRP150396. They are accessible from https://www.ncbi.nlm.nih.gov/sra/SRP150396.
Microsatellite loci were identified in 3 516 unigenes.Among these unigenes, 2 999 (85.10%) contained 1 or 2 microsatellite loci. Only 524 (14.90%) contained over 2 microsatellite loci. We identified 4 297 microsatellite loci in total. The loci had repeat motifs varying from di- to tetra-. Among them, 3 254 (75.73%) loci had dinucleotide repeat motifs. Trinucleotide repeat motifs were observed in 223 (5.2%) loci. Tetranucleotide repeat motifs were observed in 120 (2.79%) loci.

Figure 1 Locations of study sites of Hynobius chinensis in Changyang county. Sites are distinguished by the names of the corresponding localities. DNG: Dongtoutang; SHJ: Shuijingwan; JNJ: Jiangjunao; HNG: Hengtun.

Figure 2 Number of Hynobius chinensis male individuals present in a breeding pond at different dates in 2015-2016 in Dongtoutang(Changyang county, Hubei).

Table 1 Adult census population size of Hynobius chinensis estimated at different breeding ponds in different localities in Changyang County.
We developed and characterized 13 polymorphic microsatellite loci. These loci were in HWE and there were no evidence of significant LD between pairs of loci, of null alleles or genotyping errors in the 22 sample dataset. Among the 13 microsatellite loci, 7 (54%) loci have tetranucleotide repeat motifs and 6 (46.15%) have dinucleotide repeat types. The remaining locus has a trinucleotide repeat motif. The number of repeats range from 5 to 9 and the length of the loci range from 105 to 253 bp (Table 2).
The microsatellite DNA sequences have been deposited in GenBank. The characteristics and the accession number of each locus are shown in Table 2.
3.3. Genetic diversity and signature of a population bottleneckGenotype data at the 13 microsatellite loci have been obtained for all the 118 sampledH. chinensisadult individuals (see Table S1 in Appendix). As observed during the development of the microsatellite markers, the analyses in Micro-checker v2.2.3 provided no evidence of either null alleles or genotyping errors. And, there were no cases of linkage disequilibrium among the loci.Among the 13 loci, the locusHC64revealed a relatively lowPIC(Table 2) and 5 loci, i.e.HC23,HC47,HC54,HC85andHC116significantly deviated from HWE, after Bonferroni correction (P< 0.0042). All these 5 loci and the locusHC64were therefore removed from further analyses. As a result, 7 microsatellite loci (Table 3) were used in the analyses.
For the 118 samples and 7 loci, 15 different alleles were identified, resulting in 2.14 alleles per loci or averageAof 2.14 (Table 3). In other terms, the value ofAwas 2 for 6 (85.71%) out of the 7 loci (Table 3). Allele frequency ranged from 0.013 to 0.87. The maximum number of alleles (46.15%) was observed for the frequency range of 0.4-0.6 (Figure 4). 23.07% of alleles were found in the lowest frequency range, i.e. 0-0.2 (Figure 4). AverageHoandHewere 0.38 and 0.40, respectively. The average value of the inbreeding coefficient,Fiswas -0.06 (Table 3).
The value ofGWvaried between loci, ranging from 0.17 to 1, and with an average value of 0.61 (Table 3).Comparison of each locus’GWto the critical value for rejecting the null hypothesis of demographic stability(i.e. 0.68) showed thatGWwas below this value for 4(57%) loci (Table 3). The Wilconxon signed-rank test of heterozygosity excess yielded significant (One tail for heterozygosity excess) results (P< 0.05) as well as under IAM (P= 0.004), SMM (P= 0.004) and STM(P= 0.027). In the Sing test, heterozygosity excess was detected for all the 7 loci under IAM. Under TPM and also under SMM, it was detected for 5 loci namelyHC19,HC28,HC33,HC103andHC114. A significant distortion of the typical L-shaped distribution of allele frequency of putatively stable populations was detected. The historicalNewas estimated at 234 individuals. The contemporaryNewas found to be 36 (95% CI: 13.1-124.6) individuals.

Figure 3 Distribution of unigenes by size range in the transcriptome of Hynobius chinensis.
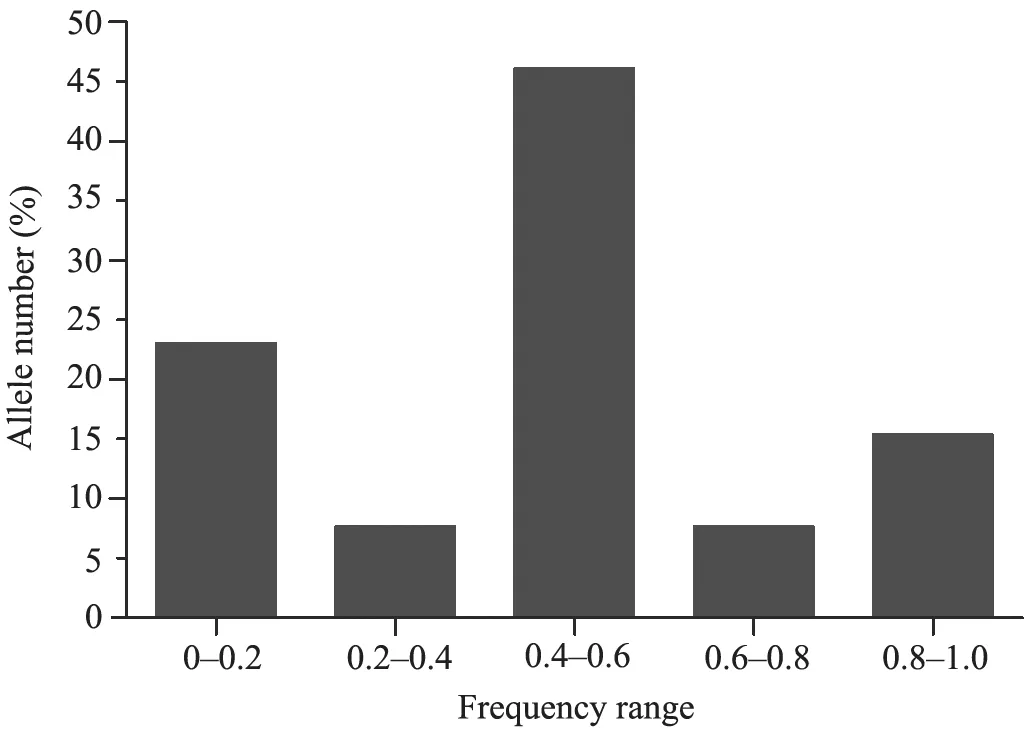
Figure 4 Distribution of alleles at seven microsatellite loci by frequency range in Hynobius chinensis (N = 118).
4. Discussion
With an average value of 0.61, the results of Garza-Williamson test indicate that the studied population ofH.chinensishas experienced a severe reduction in size or population bottleneck. The results of Wilconxon signedrank test and Mode shift test also evidence signatures of a population bottleneck.
Detection of a recent population bottleneck inH.chinensisseems to not a surprising finding (Fei and Ye, 2016; Wanget al., 2007). Literature shows thatH.chinensismay have experienced a severe demographiccrisis in the last two centuries (Wanget al., 2007).For example, Wanget al. (2007) reported that after its description at the end of the 19thcentury,H. chinensisbecame irretrievable till 2005 in the current location of the Yichang city (Wanget al., 2007).
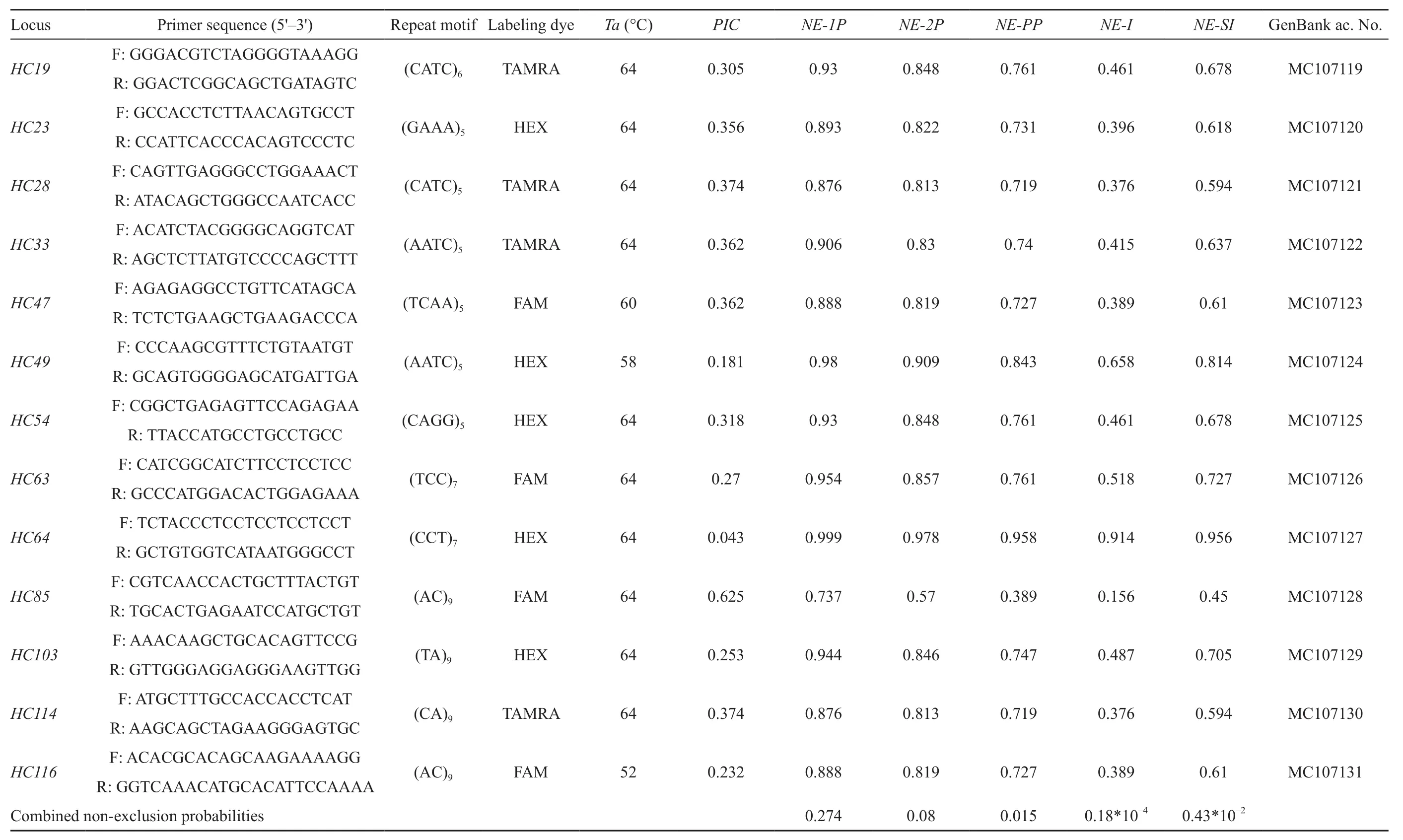
Table 2 Characteristics of thirteen novel microsatellite markers for the Chinese salamander, Hynobius chinensis.

Table 3 Values of genetic diversity indices and Garza-Williamson statistics (N = 118)
With no more than 2.14 alleles per locus, and with 0.38 and 0.40 as respective average values ofHoandHe, the genetic diversity revealed byH. chinensisseems obviously low in comparison to that reported in Least Concern (IUCN, 2018) pond-breeding Hynobiid salamanders (Matsunamiet al., 2015; Yoshikawaet al.,2013). For examples, the study of small samples from 2 populations (N= 16 andN= 8) revealed 6.63 and 3.53 alleles per locus inH. nebulosus(Yoshikawaet al.,2013). Another study of small samples (N= 20) from two populations ofH. retardatusfound that the average numbers of alleles per locus were 4.25 and 3.83. The averageHofor these populations was 0.64 and 0.57; and the averageHewas found to be 0.59 and 0.49 (Matsunamiet al., 2015).
The level of genetic diversity revealed byH. chinensisin the present study seems close to that reported in other threatened and narrowly distributed (IUCN, 2018) closely related species such asH. dunni(Sugawaraet al., 2015),H. maoershanensis(Linet al., 2015),and H. tokyoensis(Sugawaraet al., 2015). A recent study of 12 populations ofH. dunnireported an averageAof 2.53 and 0.34 as the value ofHoandHe(Sugawaraet al., 2015). In a another recent large-scale study ofH. tokyoensis, a forestdwelling Hynobiid salamander threatened by habitat loss and fragmentation due to land use changes as it might be in the focal species (Fei and Ye, 2016) which involved 46 populations from 12 different geographic regions,Sugawaraet al. (2016) found comparable results. These authors showed that the maximum average value ofAwas 3.4. They also showed that averageAwas < 2.0 in 39.13% of populations and between 2.0 and 2.40 in 32.61% of populations (Sugawaraet al., 2016).Heranged from 0.02 to .51 and averaged 0.27 for all the populations.
Our demographic and genetic diversity results seem to corroborate findings of earlier studies, using microsatellite data, which report population bottlenecks (Apodacaet al.,2012; Heredia-Bobadillaet al., 2017; Wanget al., 2017),and levels of genetic diversity similar to that revealed byH. chinensisin this study (Jordanet al., 2009; Spearet al.,2006) in a variety of threatened salamanders, including Ambystomatids, and Phlethodontids.
Inbreeding is one of the mechanisms expected to lead to loss of genetic diversity after population bottlenecks in threatened and narrowly distributed amphibians (Ficetolaet al., 2011; Storferet al., 2014). However, with an average value of -0.06, the inbreeding coefficient,Fisprovides evidence of moderate level of inbreeding in the present study.
Analyses of microsatellite data in Genepop v1.2 software detected departures from HWE for 5 microsatellite loci (i.e.HC23,HC47,HC54,HC85andHC116). These departures cannot be attributed to null alleles because a significant occurrence of such alleles in the dataset has not been confirmed by analyses in Micro-Checker v2.2.3. In comparison to loci in HWE, these loci exhibited heterozygosity deficiency (Results not shown).Hence, though inbreeding coefficientFisestimated without these loci revealed a moderate level of inbreeding, the departures from HWE can be considered as a consequence of inbreeding following the inferred population bottleneck(Apodacaet al., 2012; Storferet al., 2014). Inbreeding may have caused loss of heterozygosity, which resulted in the observed low level of heterozygosity (Storferet al.,2014). The level of inbreeding may have been brought at a moderate level by selection against inbreeding (Ficetolaet al., 2011) and by polyandry (Slatyeret al., 2012). Few inbreed individuals may reach maturity (Ficetolaet al.,2011). During this study, at all observation opportunities(8 in total), we found that an individual female’s eggs were seemingly fertilized by several males (2-7). Such a mating pattern may result in a reduced probability of inbreeding (Slatyeret al., 2012).
The low number of alleles revealed byH. chinensisin this study may have been a consequence of loss of rare alleles by genetic drift following the inferred population bottleneck (Garza and Williamson, 2001). Loss of such alleles may have been facilitated by fragmentation of populations caused by forced exclusion ofH. chinensisfrom proportions of natural habitats (Allentoft and O’Brien, 2010; Apodacaet al., 2012; Sugawaraet al.,2016). Also, a low density of breeders (i.e. 386-404 breeders per 2.18 km2) may have contributed to fragmentation of populations (Allentoft and O’Brien,2010).
According to interviewed people, environmental conditions have considerably changed in the study area since 1960s. Since that time period, the local forest that is also the habitat ofH. chinensiswas intensively harvested.From valleys toward mountain tops, cultivated lands expanded rapidly, reaching almost the current area in 1970s. The forest rapidly regressed towards mountain tops and became fragmented. As larger trees were selectively cut, the density of the canopy rapidly decreased and the forest become degraded. Cultivated lands rapidly became infertile, and the use of fertilizers became intensive in 1970s.
Interviewed people indicated that fire was largely used to clear forest areas for land cultivation, suggesting a possible massive and repeated decimation of terrestrial life stages ofH. chinensis. Human-induced forest reduction, fragmentation and degradation were evident in all the study localities. Exposure ofH. chinensis(both aquatic and terrestrial stages) to agrochemicals was also suspected.
These human-induced changes in the potential habitat may have affected gene flow and population growth rate and caused population fragmentation and declines,triggering loss of alleles and heterozygosity inH.chinensis(Allentoft and O’Brien, 2010; Apodacaet al.,2012; Heredia-Bobadillaet al., 2017; Rivera-Ortízet al.,2015; Sugawaraet al., 2016).
In addition, interviewed people revealed that during breeding seasons, adultH. chinensisindividuals are collected from breeding ponds and used as a source of proteins. This finding suggests that overharvesting may be another contributing factor to population declines and loss of genetic diversity (Hauseret al., 2002) in the study system.
The values of adult census population size,Ncrevealed byH. chinensisin the present study appears to be smaller than values reported by earlier studies of close related species under suitable habitat conditions (Fuet al., 2003;Guet al., 1999; Ma and Gu, 1999), suggesting a possible reduced abundance ofH. chinensisin the study area due to threats mentioned above (Chenet al., 2016; Fei and Ye, 2016). For example, survey data fromH. amjiensisgathered from 1992 to 1998 (Guet al., 1999), fromH.yiwuensis-identified by the authors asH. chinensisgathered in 1985, 1988 and 1998 (Ma and Gu, 1999) and personal observation ofH. yiwuensisby Fuet al. (2003)show that in natural and less anthropogenically disturbed habitats, the number of female breeders per breeding pond should exceed 50, even 100 individuals (Chenet al.,2016; Fuet al., 2003; Guet al., 1999; Ma and Gu, 1999).
Comparison of the contemporaryNeto the historicalNeshows that the contemporaryNeis more than 6 orders of magnitude smaller than the historicalNe, evidencing a long-term reduction in population size. This suggests thatH. chinensismay have undergone population declines and loss of genetic diversity prior to the anthropogenic threats listed above (Jordanet al., 2009). And, long-term declines may have also significantly contributed to fragmentation in gene pools and loss of genetic diversity (Jordanet al.,2009).
We report data onNcinH. chinensisfrom three different localities collected over three successive breeding period. Despite a relative stability during the study period, asNcmay naturally highly fluctuate, longterm data may be required to know the exactNcin the study localities (Beebee and Griffiths, 2005; McCartney-Melstad and Shaffer, 2015). The observed variability inNcbetween localities suggests that local factors may govern the distribution and abundance ofH. chinensis(Beebee and Griffiths, 2005; Guet al., 1999). And, this should also be true for genetic diversity (Beebee and Griffiths,2005; Sugawaraet al., 2016). Evidence from closely related species such asH. amjiensisshows that variation inNcmay be caused by spatial variation in humaninduced changes in ponds’ water quality, and physical characteristics (Guet al., 1999).
We sampled the largest population found, i.e.Taungtotang’s population expecting to see a better genetic variability and demographic history (Hoffmannet al.,2017). But our results show that despite its bigger size,this population is genetically poor and bottlenecked.And, as expected for amphibians (Allentoft and O’Brien,2010; Beebee and Griffiths, 2005),Newas several orders of magnitudes smaller thanNc. Note that, in 2015, we recorded 140H. chinensisadult individuals in Taungtotang, resulting in aNe/Ncratio of 0.26 (or 36:140). Though literature shows that higher values can be reached in some species (e.g.Georcrina vitellina,Rana sylvatica), the value of this ration is < 0.2 in many amphibians, including anurans and Caudata (Beebee and Griffiths, 2005). AsNcwas smaller in the other localities than in Taungtotang, this relationship suggests thatNeand genetic diversity must be more reduced in those localities(Hoffmannet al., 2017), suggesting a possible extirpation ofH. chinensis’ populations.
As mentioned above, information on landscape use lacks forH. chinensis. The results of this study demonstrate the paucity of the existing information. For example, while expected to occur at altitudes ranging from 1400 to 1500 m (Fei and Ye, 2016), we found two (50%) breeding populations, including the largest breeding populations beyond this range, at altitudes of 1548 m and 1582 m precisely. The lack of such information prevents us from appreciating the sampling effort of this study and hence the significance of the results at the species level (Allentoft and O’Brien, 2010;Beebee and Griffiths, 2005).
In conclusion, this study makes available thirteen novel microsatellite markers forH. chinensis. It provides information on the exact location of populations and breeding sites. This study provides molecular evidence of a population bottleneck inH. chinensis, which is provided by the Garza-Williamson test, Heterozygosity excess tests, Mode shift test and estimates of effective population sizes. It shows thatH. chinensismay exist in small-sized populations, which retain low microsatellite genetic diversity. Together, our results suggest that populations ofH. chinensismay have been extirpated in the study area.
For further understanding of the demography and genetic viability ofH. chinensis, future studies should also investigate the distribution and sizes of the representative populations of this endangered species. They should investigate fitness effects of inbreeding and specific causes of population declines while considering gene flow and populations genetic structuring. Conservation initiatives should aim to conserve and restore the habitats and regulating the exploitation of the species. They should aim to maintain the extant genetic variation.
AcknowledgementsThe authors want to thank A.STOJKOSKA, who revised the manuscript. They also express their gratitude to Y. LIU and to S. LIU from Dongtoutang, who helped collect information on the evolution of landscapes and on the usage of the focal species by humans in the study area. This study was financially supported by the Chinese National Science and Technology Basic Work Program (No. 2014FY110100)and the State Forestry Administration of China.
Appendix
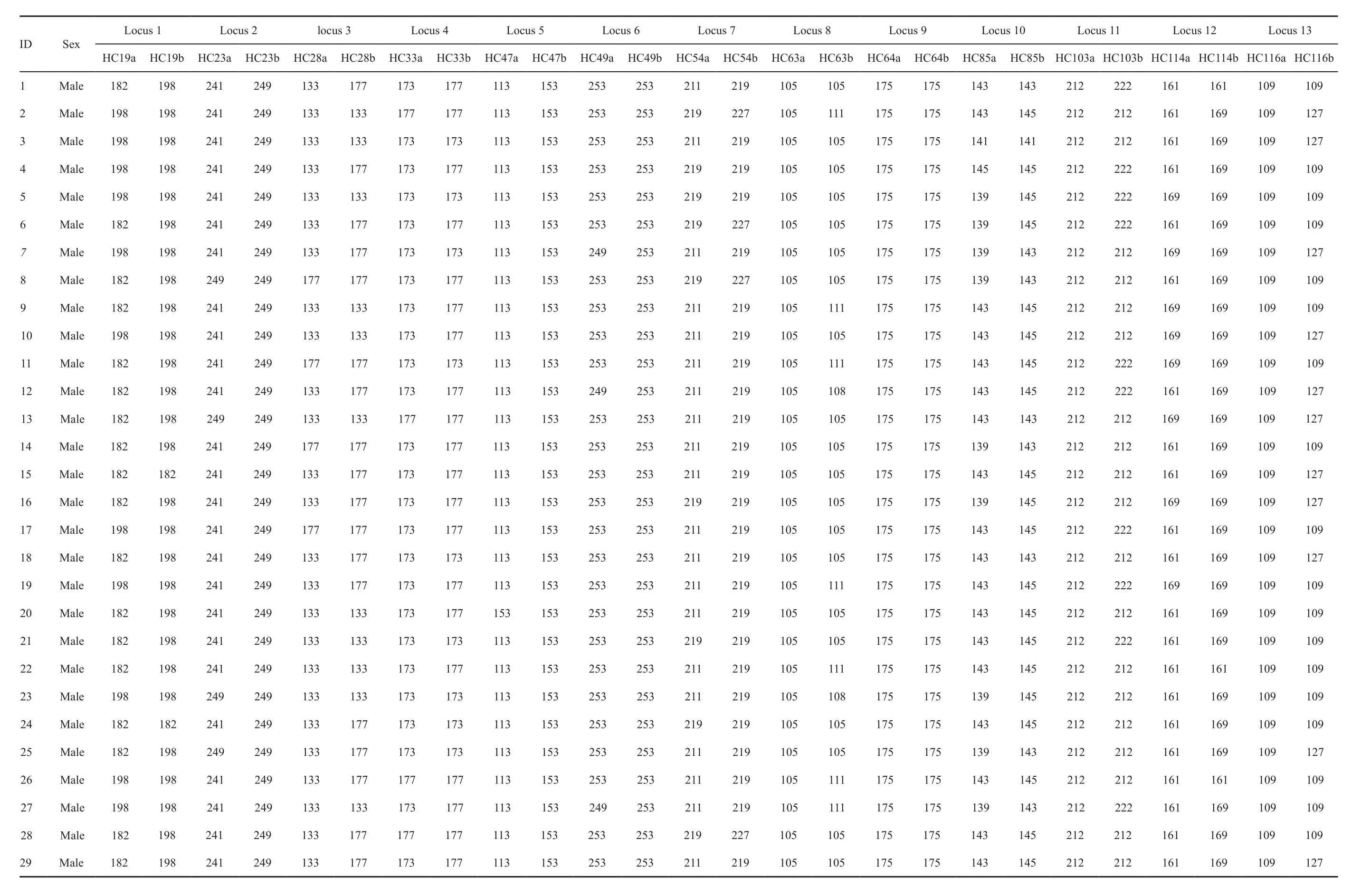
Table S1 The microsatellite genotype data (13 loci) from adults individuals of Hynobius chinensis used (N =118)

Continued Table S1
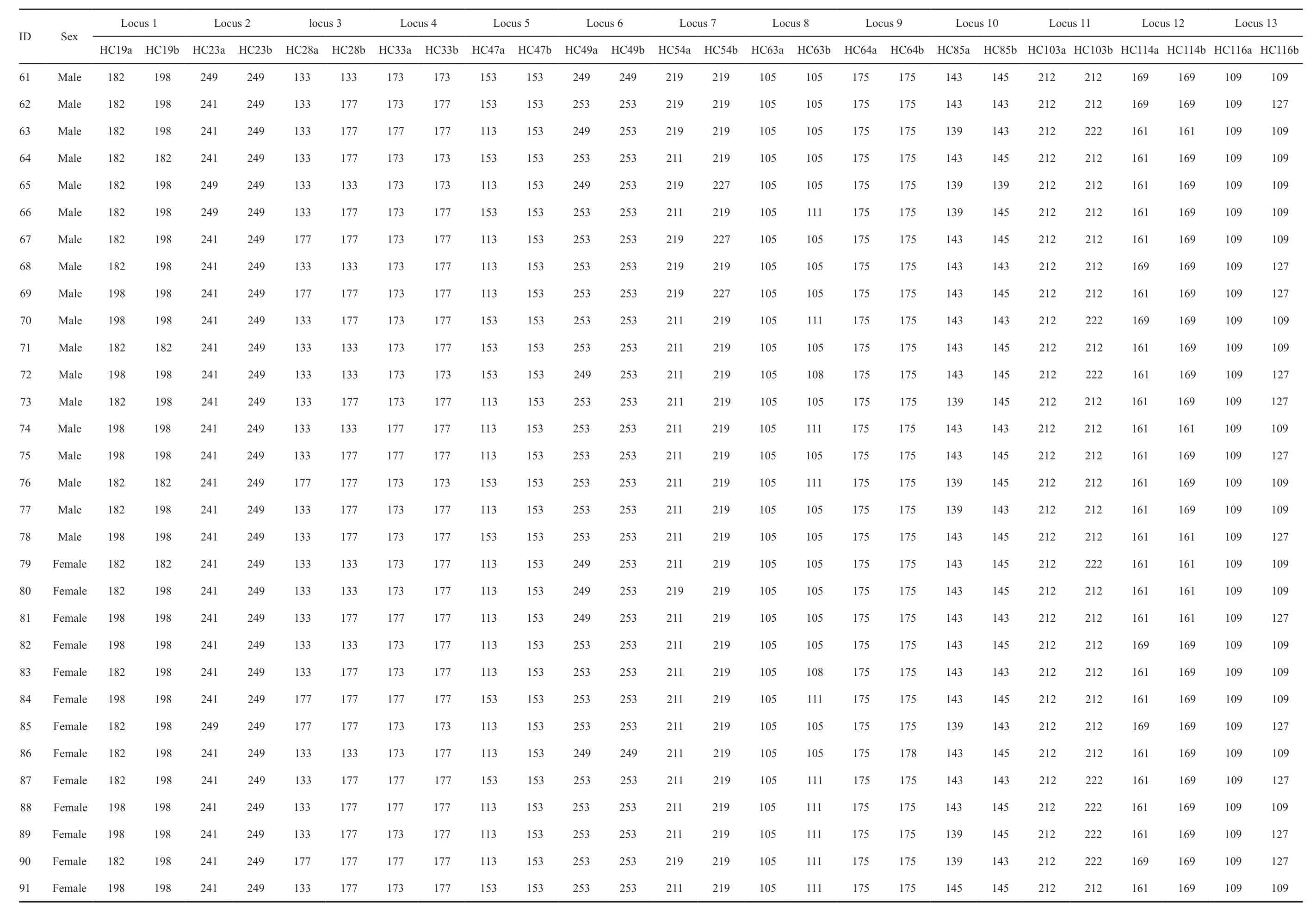
Continued Table S1

Continued Table S1
 Asian Herpetological Research2018年3期
Asian Herpetological Research2018年3期
- Asian Herpetological Research的其它文章
- A New Species of Genus Microhyla (Amphibia: Anura:Microhylidae) from Zhejiang Province, China
- A Review for Life-history Traits Variation in Frogs Especially for Anurans in China
- Perch Associated Expression of Phenotypic Plasticity in Limb Development and Sprint Speed in Agamid Lizard Calotes versicolor: A Laboratory Study
- Tail Waving Speed Affects Territorial Response in the Toad-headed Agama Phrynocephalus vlangalii
- Predatory Cues Influence the Behavioral Responses and Metamorphic Traits of Polypedates maculatus (Anura:Rhacophoridae)
- Massive Molecular Parallel Evolution of the HSP90AA1 Gene between High-elevation Anurans