
伊枯草菌素A发酵过程中游离氨基酸的HPLC分析
2018-09-08 07:56赵杰岳华苟学磊周金燕谭红
生物技术通报 2018年8期
赵杰 岳华 苟学磊 周金燕 谭红
(1. 中国科学院成都生物研究所 中国科学院环境与微生物重点实验室,成都 610041;2. 中国科学院大学,北京 100039)
伊枯草菌素A(Iturin A)最初是从枯草芽孢杆菌(Bacillus subtilis)发酵液中分离得到的一种环脂肽类抗生素,其结构由一个β-脂肪酸侧链和7个α-氨基酸残基(L-Asn-D-Tyr-D-Asn-L-Gln-LPro-D-Asn-L-Ser-)构成[1,2]。具有强烈的抗真菌活性[3],并且低毒、低致敏性,能抑制西瓜枯萎病菌(Fusarium oxysporum)[4]、南部玉米叶斑病原菌(South corn leaf blight)[5]、禾谷镰孢菌(Fusarium graminearum)[6]等多种植物病原真菌的生长,还可用于人和动物皮肤真菌病的治疗[7]。因而,在农业和医药方面具有较好的应用前景及潜在的研究价值。
近年来,国内外对iturin A的发酵工艺进行了大量研究,Hbid等[8]认为较高溶氧量有利于iturin A的产生。Shih等[9]利用响应面法优化枯草芽孢杆菌 S3的发酵条件,使iturin A的产量达11.44702 mg/g固体湿重。金虎等[10]利用未经处理的菜籽粉作为氮源,并以一种新型两步葡萄糖补料方法生产iturin A,使iturin A产量达到1.12 g/L。彭文璟等[11]基于均匀设计的人工神经网络模型,优化iturin A的补料分批发酵条件,将iturin A效价优化至13364.5±271.3 U/mL。这些研究虽然使iturin A产量得到一定的提高,但依旧未能实现工业化生产。另有研究表明,iturin A是经非核糖体途径合成的环脂肽抗生素,其合成过程需要结合氨基酸以完成结构转化[12]。在iturin A合成代谢途径中,Asn、Val可作为iturin A的重要合成前体,对iturin A的合成具有促进作用[13-14],并且培养基中氨基酸的种类和含量对iturin A同系物的组成比例有影响[15]。氨基酸既可作为iturin A的结构组分,又可作为iturin A合成酶的诱导物,在iturin A生物合成过程中起着关键性作用。因此,分析检测iturin A发酵过程中各种游离氨基酸,可以明确培养基中氨基酸的种类、含量以及被利用的时间和速度,对人工有效调控发酵过程中氨基酸浓度来提高iturin A产量具有重要意义。
为快速、精确研究iturin A发酵过程中游离氨基酸含量的变化,建立一种适合此样品中游离氨基酸的测定方法至关重要。目前测定氨基酸的方法主要有氨基酸分析仪法[16]、毛细管电泳法[17]和高效液相色谱法[18]等。氨基酸分析仪因设备昂贵、用途单一导致其使用受限。任艳丽等[19]报道用毛细管电泳法测定枯草芽孢杆菌发酵液中游离氨基酸含量,但此方法稳定性及重现性差,无法对His、Arg、Ile和Leu进行准确的定性和定量分析。而高效液相色谱(HPLC)结合柱前衍生的方法具有灵敏度高、适用性广、且能同时准确测定多种氨基酸等特点[20],并且液相色谱仪带有自动进样系统,是一种较为理想的氨基酸检测方法。柱前衍生HPLC法使用的关键在于衍生试剂和液相色谱条件的选择。本研究拟通过采用在室温下能和氨基酸发生反应,且衍生产物PTC-AA成分单一稳定,衍生副产物对测定无干扰的异硫氰酸苯酯(PITC)[21]为柱前衍生剂,优化HPLC检测条件,以建立一种iturin A发酵液中游离氨基酸的快速检测方法,提高各氨基酸的分离度,为研究iturin A发酵过程中游离氨基酸的代谢规律及调控发酵过程中氨基酸浓度提供技术支持。
1 材料与方法
1.1 材料
1.1.1 菌种 枯草芽孢杆菌(Bacillus subtilis)ZK8,由本实验室从产自新疆阿克苏的棉花植株上分离纯化、诱变而得。
1.1.2 试剂 混合氨基酸标准溶液:天门冬氨酸(Asp)、谷氨酸(Glu)、丝氨酸(Ser)、甘氨酸(Gly)、组氨酸(His)、精氨酸(Arg)、苏氨酸(Thr)、丙氨酸(Ala)、脯氨酸(Pro)、酪氨酸(Tyr)、缬氨酸(Val)、蛋氨酸(Met)、异亮氨酸(Ile)、亮氨酸(Leu)、苯丙氨酸(Phe)、赖氨酸(Lys)。除半胱氨酸(Cys)浓度为1.25 μmol/mL外,其余16种氨基酸浓度均为2.5 μmol/mL,内标物质为正亮氨酸(Nle)、异硫氰酸苯酯(PITC)以及三乙胺(TEA),以上试剂均购自天津博纳艾杰尔科技有限公司;色谱级乙腈,美国霍尼韦尔公司;无水乙酸钠,天津市科密欧化学试剂有限公司;冰醋酸、甲醇,天津市博迪化工股份有限公司;水为超纯水。
1.1.3 仪器 Agilent 1260高效液相色谱仪,美国Agilent公司;5415R高速冷冻离心机,德国Eppendorf公司;BUCHI旋转蒸发仪R-300,瑞士步琦有限公司;电子分析天平,上海梅特勒—托利多仪器有限公司;pH计,赛多利斯科学仪器有限公司;旋涡混合器购自上海青浦沪西仪器厂。
1.2 方法
1.2.1 溶液配制 系列混合氨基酸标准溶液:取氨基酸混合标准溶液,用超纯水将其稀释为0.1、0.5、0.9、1.3、1.7、2.1、2.5 μmol/mL 7个浓度梯度标准溶液;三乙胺乙腈溶液:取三乙胺1.4 mL,加乙腈8.6 mL,混匀;异硫氰酸苯酯乙腈溶液:取异硫氰酸苯酯25 μL,加乙腈2 mL,混匀(密闭,4℃保存);正亮氨酸内标溶液:精密称取正亮氨酸10 mg,加0.1 mol/L 盐酸溶液10 mL溶解,混匀。
1.2.2 衍生方法 氨基酸衍生参照文献[22]方法,略作修改。准确量取混合氨基酸标准溶液及样品溶液200 μL,分别置于1.5 mL 的EP管中,依次加入20 μL正亮氨酸内标溶液、100 μL三乙胺乙腈溶液及100 μL异硫氰酸苯酯乙腈溶液,摇匀,室温静置衍生1 h。衍生结束后加入400 μL正己烷,震荡摇匀,静置10 min,取下层溶液(PITC-AA)200 μL,用600 μL超纯水稀释,混合均匀后用0.45 μm针式过滤器过滤。
1.2.3 氨基酸色谱分析条件优化 色谱柱:Venusil AA氨基酸分析专用柱(4.6 mm ×250 mm,5 μm);流动相A:0.1 mol/L无水乙酸钠-乙腈溶液(66:5);流动相B:80%乙腈水溶液;流速:1 mL/min;检测波长:254 nm;进样量:10 μL;优化其余3个色谱条件。
1.2.3.1 流动相梯度洗脱程序的优化 以文献[23-24]中的梯度洗脱程序为基础,并结合所测定各氨基酸的极性特性,多次调节流动相A、B的比例,使其随洗脱时间连续发生改变以此连续改变流动相的极性,考察不同梯度洗脱程序对混合氨基酸分离效果的影响,以确定能在最短时间内实现氨基酸最佳分离的梯度洗脱程序。
1.2.3.2 流动相pH值的优化 配制0.1 mol/L无水乙酸钠溶液4份,用冰醋酸调节pH值分别为4.0、5.0、6.0、7.0,加入一定体积乙腈,使无水乙酸钠与乙腈的体积比为66∶5,过0.45 μm 滤膜,得到不同pH值的流动相A。其余色谱条件不变,混合氨基酸标准溶液经HPLC在不同pH值的流动相中洗脱测定,考察流动相pH值对各氨基酸分离效果的影响,以确定最佳的流动相pH值。
1.2.3.3 色谱柱温的优化 分别设置色谱柱温度为25℃、30℃、35℃、40℃,其余色谱条件不变,混合氨基酸标准溶液经HPLC在所设定的色谱柱温度下测定分析,考察色谱柱温对各氨基酸分离效果的影响,以确定最佳的色谱柱温度。
1.2.4 方法验证
1.2.4.1 线性关系 将“1.2.1”所配制的系列混合氨基酸标准溶液按“1.2.2”项方法衍生处理后,在优化后的色谱条件下经HPLC检测分析,以氨基酸峰面积与内标Nle峰面积之比为纵坐标,以氨基酸浓度(mg/L)为横坐标,绘制标准曲线,进行线性回归分析,并计算相关系数(R2)。
1.2.4.2 精密度及稳定性 取浓度为2.1 μmol/mL氨基酸混合标准溶液,按“1.2.2”项中的衍生方法衍生处理,在优化后的色谱条件下连续进样6次,计算氨基酸峰面积与内标峰面积之比以及保留时间的相对标准偏差(RSD),考察方法精密度;另对同一浓度氨基酸混合标准溶液衍生处理,并于衍生处理后0、2、4、6、8、12、18、24 h依次进样测定,计算氨基酸峰面积与内标峰面积之比以及保留时间的RSD值,考察方法稳定性。
1.2.4.3 重复性 精确量取发酵0 h的iturin A发酵液6份,经预处理及衍生后,分别进样测定,计算每种氨基酸浓度和RSD值。
1.2.4.4 加标回收率 精确量取已知浓度的iturin A发酵液6份,分别加入一定浓度混合氨基酸标准品,经样品预处理及衍生后,进行HPLC测定分析,计算回收率。
1.2.5 发酵液制备
1.2.5.1 发酵培养基 葡萄糖 42.6 g/L,大豆蛋白胨总氮 2.0 g/L,酵母膏 0.12 g/L,MgSO4·7H2O 3.14 g/L,KH2PO43.62 g/L,pH 8.0,121℃灭菌 30 min。
1.2.5.2 摇瓶分批发酵 取一支生产良好的枯草芽孢杆菌ZK8斜面,转接于种子培养基中(装样量为100 mL/500 mL三角瓶),150 r/min、30℃条件下震荡培养20 h。然后将种子液按10%转种量转种至发酵培养基中,150 r/min、30℃条件下震荡培养,并于发酵开始后不同时间点取样,取样时间点为0、3、6、9、12、15、18、24、28、32、36、42、48、54、60 h。样品于12 000 r/min离心10 min,取上清液,冷冻保存备用。
1.2.6 样品预处理 取250 μL发酵上清液,加入1 mL无水甲醇,摇匀,静置1 h,12 000 r/min离心10 min,吸取1 mL上清液用旋转蒸发仪于50℃水浴蒸干,最后加200 μL超纯水溶解备用。
2 结果
2.1 色谱条件优化
2.1.1 梯度洗脱程序优化 HPLC 色谱图结果(图1)显示,不同梯度洗脱程序对混合氨基酸分离时间、分离效果有显著影响。当流动相B(80%乙腈)初始比例为0%,在2-15 min增加至10%、15-25 min增加至30%、25-33 min增加至45%、33-33.1 min增加至100% 时,可在35 min内将所有氨基酸完全分离(图1-c),与条件a(图1-a)相比,氨基酸在色谱柱中的保留时间缩短了20 min,且Pro与杂质峰得到有效分离,故最优梯度洗脱程序见表1。
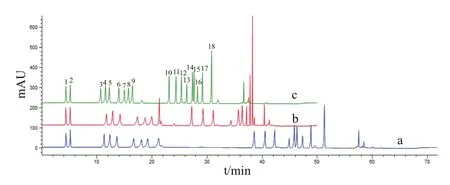
图1 不同梯度洗脱程序下混合氨基酸标准品色谱图
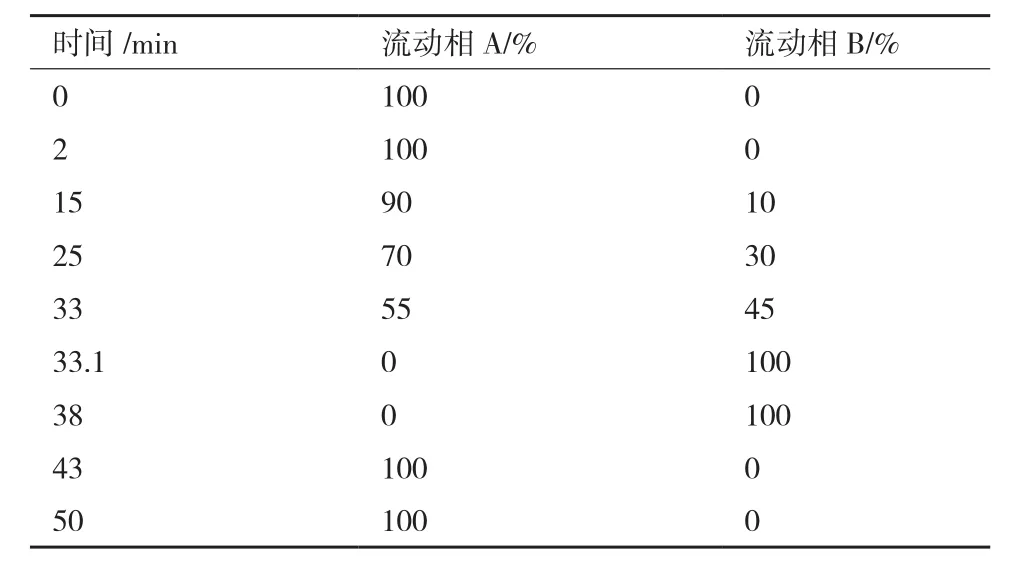
表1 梯度洗脱程序
2.1.2 流动相pH值优化 HPLC在不同pH值的流动相中分离混合氨基酸的结果(图2)显示,当流动相pH值为4.0、5.0时,氨基酸色谱峰重叠现象严重,分离效果较差;当流动相pH值为6.0、7.0时,所有氨基酸完全分离,并且分离度均大于1,仅Ser、Gly、His、Arg、Thr、Ala和Pro色谱峰有前延现象。在pH值6.0-7.0之间进一步实验后确定流动相pH值为6.4±0.1。
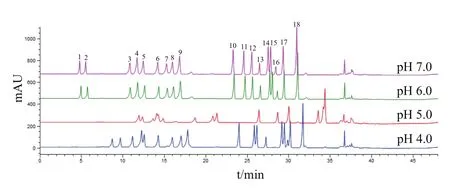
图2 不同流动相pH值下氨基酸标准品色谱图
2.1.3 色谱柱温优化 HPLC在不同色谱柱温度下分离混合氨基酸的结果(图3)显示,升高色谱柱温有利于目标物的分离,当色谱柱温上升至40℃时,各氨基酸色谱峰无明显前延、重叠现象,峰型较好,且分离度均大于1.5,因此选择40℃为最终柱温。

图3 不同色谱柱温下氨基酸标准品色谱图
2.2 方法验证
2.2.1 线性关系 将系列浓度混合氨基酸标准溶液衍生处理后,在优化后的色谱条件下经HPLC检测,计算分析得到各氨基酸线性回归方程、相关系数、线性范围,结果见表2。16种氨基酸在一定浓度范围内具有良好的线性关系,但半胱氨酸衍生物不稳定易分解,因而只对其进行了定性分析。
2.2.2 精密度及稳定性 按照实验方法测定并计算,结果见表3,各氨基酸峰面积与内标峰面积之比以及保留时间的RSD值均在2%以内,表明该方法波动小、精密度高,且16种氨基酸衍生物在24 h内均具有较好稳定性。

表2 16种氨基酸的线性关系
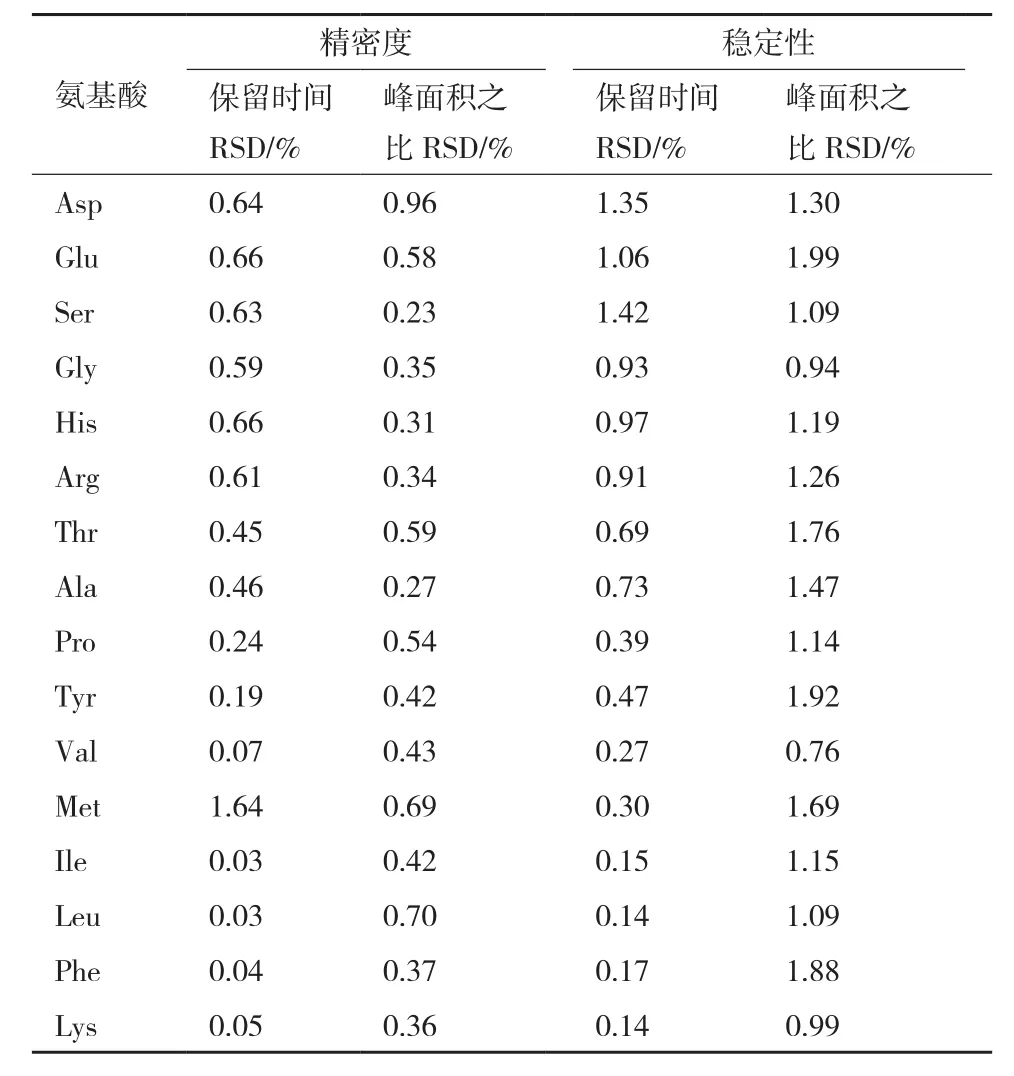
表3 精密度和稳定性验证结果
2.2.3 重复性 取0 h的iturin A发酵液6份,经预处理及衍生后分别进样1次,结果见表4,各氨基酸浓度的RSD值均小于5%,表明该方法的重复性良好。
2.2.4 加标回收率 16种氨基酸的加标回收率在83.84%-108.02%之间,且RSD值均小于2.77%,见表4,表明该测定方法准确性高,满足分析要求。
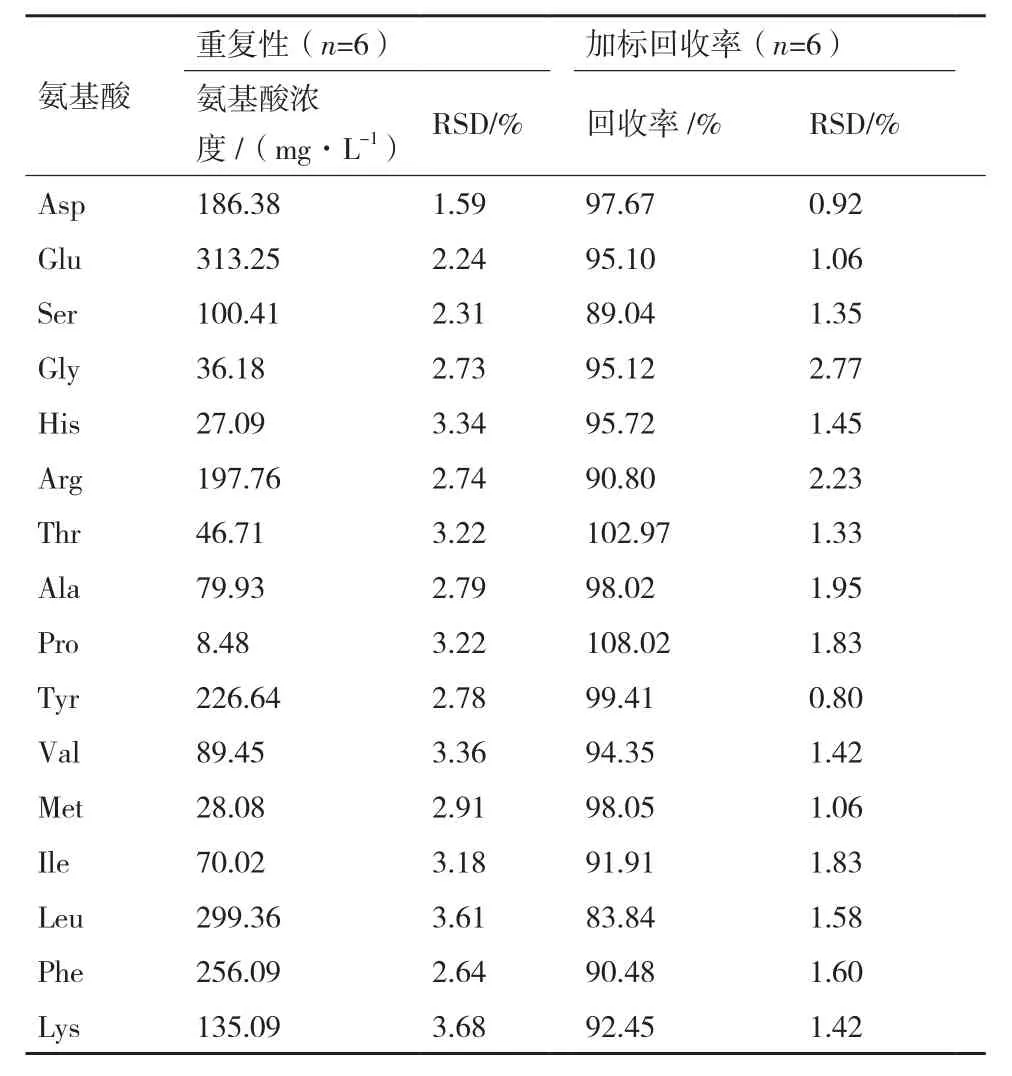
表4 重复性和加标回收率结果
2.3 伊枯草菌素A发酵液中游离氨基酸的动态变化分析
Iturin A发酵液样品在优化后的色谱条件下经HPLC测定分析,得到样品中各游离氨基酸的分离图谱,见图4。用内标曲线法计算各时间点样品中16种氨基酸浓度,得到iturin A发酵过程中16种游离氨基酸浓度随发酵时间的变化规律,见图5。
由图5-A知,游离氨基酸总含量呈一个游离氨基酸被消耗与释放的动态变化过程。在发酵0-15 h期间,发酵液中游离氨基酸的总含量随着时间增加而降低,可能原因为此阶段菌体生长较快,游离氨基酸被枯草芽孢杆菌大量消耗用于菌体生长,18 h后其总含量呈较明显的上升趋势,推测可能与培养基中多肽、蛋白质不断被分解为氨基酸有关。Iturin A于发酵3 h开始合成,随后产量逐渐增加直至发酵结束。
16种游离氨基酸初始浓度各不相同,且在发酵过程中呈现出不同的变化趋势。其浓度变化趋势大致可分为三类:第一类为游离Tyr,其浓度在整个发酵过程中变化不大,见图5-B。第二类为游离Asp、Glu、Ser、Arg、Ala、Leu、Gly、Thr、Met, 在 发酵过程中其浓度均呈下降趋势。其中,Asp、Leu、Glu、Gly这4种氨基酸浓度在发酵0-24 h期间逐渐下降,Ser、Ala则于0-15 h间处于下降阶段,之后其浓度均无明显变化,Arg、Thr浓度在整个发酵过程中缓慢下降,Met在发酵0-28 h期间浓度迅速下降,之后其浓度小幅升高,见图5-C。第三类是游离 His、Pro、Val、Ile、Phe、Lys,发酵过程中其浓度呈上升趋势,其中Val的上升速率最大,见图5-D。
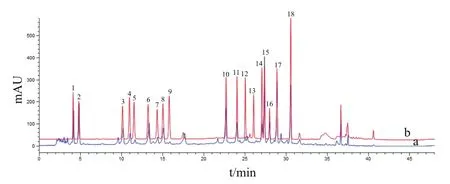
图4 氨基酸标准品及样品色谱图
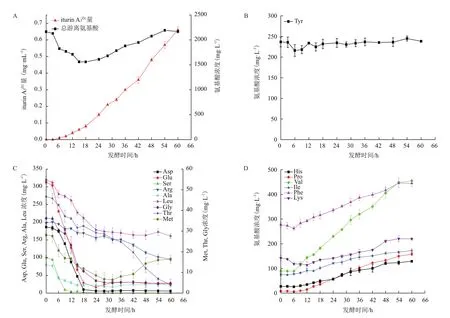
图5 枯草芽孢杆菌发酵过程中游离氨基酸及iturin A合成代谢曲线
3 讨论
在微生物发酵产生抗生素过程中,氨基酸与抗生素合成密切相关,且在发酵过程中添加氨基酸能促进抗生素合成[25-27]。Iturin A作为环脂肽类抗生素,已有研究表明氨基酸对其生物合成存在影响。Besson等[28,13]考察了14种氨基酸分别作为发酵培养基中唯一氮源对iturin A合成的影响,发现结构骨架中的L-Asn是合成iturin A的最佳底物,而在L-Pro和D-Tyr中不能合成iturin A,后期采用同位素标记α-氨基酸(Asn、Gln、Ser、Pro和Tyr)进一步研究iturin A的生物合成,表明L-Asn可能是iturin A合成的最好前体。Hourdou等[14]研究发现Val是iturin A侧链脂肪酸部分合成的前体。Iwase等[15]通过向以脱脂豆粉为氮源的培养基中添加不同种类氨基酸研究发现氨基酸种类对iturin A同系物的组成比例有影响。许锬等[29]和彭文璟等[11]研究表明在枯草芽孢杆菌发酵过程中添加Asp、Glu、Pro能促进iturin A的合成,并采用人工神经网络(ANN)方法优化这三种氨基酸在分批发酵中的补料浓度,使iturin A的效价达13364.5 ± 271.3 U/mL,较对照提高了34.6%。由此可见,氨基酸对iturin A的生物合成具有重要影响。采用高效、便捷的检测手段监测分析发酵过程中各种游离氨基酸含量的动态变化有助于深入了解iturin A的合成代谢,并可为调控iturin A发酵过程中氨基酸的浓度,提高iturin A的产量奠定基础。
由于iturin A发酵液中含有多种游离氨基酸,这些氨基酸不含有芳香环等生色团无法直接用紫外检测器检测[30],并且各氨基酸的保留性质强弱不一[31],以及氨基和羧基的解离情况受到pH值及温度的影响[32]。因此,需要选用一种合适的衍生剂衍生氨基酸,在最佳的条件下实现各氨基酸的有效分离。本研究采用高效液相色谱,以异硫氰酸苯酯(PITC)为衍生剂衍生氨基酸,对梯度洗脱程序、流动相pH和色谱柱温3个色谱条件进行优化选择,使得各氨基酸在35 mim内被完全分离,缩短了分析时间,提高了分析效率。所使用的PITC衍生剂衍生得到的氨基酸衍生物除半胱氨酸衍生物外,其余均非常稳定,而且衍生步骤简单,对环境要求低。与毛细管电泳法[19]相比,PITC柱前衍生高效液相色谱法可显著改善部分氨基酸的分离度,能有效分离出的氨基酸种类(16种)比毛细管电泳(14种)法多,且准确性更高,尤其更能准确定性和定量分析His、Arg、Ile和Leu。该方法具有良好的线性关系(R2>0.996),灵敏度高、衍生产物稳定、精密度和重现性好,适合用于iturin A发酵液中多种游离氨基酸成分和含量的测定。
4 结论
本研究采用PITC将氨基酸在柱前转化为适于高效液相色谱分离及检测的衍生物,并优化了色谱条件,建立了柱前衍生高效液相色谱测定伊枯草菌素A发酵液中游离氨基酸含量的方法。该方法线性关系良好,衍生产物稳定、分析时间短、精密度高、重复性好。同时,研究分析了伊枯草菌素A发酵过程中16种游离氨基酸浓度变化规律,为发酵过程中调控氨基酸浓度提高iturin A产量提供了科学依据。
猜你喜欢
实用手外科杂志(2022年2期)2022-08-31
湖南饲料(2021年4期)2021-10-13
科学导报·学术(2020年29期)2020-10-21
西南石油大学学报(自然科学版)(2019年5期)2019-12-20
扬子江(2019年3期)2019-05-24
天然产物研究与开发(2018年4期)2018-05-07
中成药(2018年1期)2018-02-02
浙江农业学报(2017年1期)2017-05-17
现代检验医学杂志(2015年4期)2015-02-06
外语教学理论与实践(2014年2期)2014-06-21
