
手性2,2'-联吡啶衍生物的不对称合成
2018-08-28 11:20:12王广慧乔秀丽迟彩霞董立强
绥化学院学报 2018年9期
苏 适 王广慧 乔秀丽 迟彩霞 王 斌 董立强
(绥化学院食品与制药工程学院 黑龙江省绥化 152061)
肿瘤血管是肿瘤生长和转移的病理基础,血管内皮生长因子是肿瘤血管生成的重要调节因子,它与肿瘤血管的生成有非常密切联系。缺少新生血管的人体肿瘤会一般被限制在很小的范围内无法生长扩散,因此,抑制和阻断血管内皮生长因子在肿瘤组织中的表达可达到抑制肿瘤细胞的生长和转移的目的[1,2]。研究结果显示,分子中含有氮芳杂环、酰胺结构以及亚胺结构的血管内皮生长因子小分子抑制剂,在体外细胞和动物的实验中对肿瘤细胞均表现出良好的抑制作用。
本文对此类化合物构效关系分析发现,分子中的芳环或取代芳环可作为疏水性基团,分子中氮芳杂环中的氮原子、酰胺键可与血管内皮生长因子产生氢键作用[3]。因此,设计合成具有多个结合位点,生物活性高的血管内皮生长因子小分子抑制剂已经成为抗肿瘤药物研究的热点。基于对血管内皮生长因子抑制剂构效关系分析,设计了以吡啶衍生物为起始原料,通过缩合反应引入不同保护基和不同构型的光学纯的α-氨基酸作为手性源来调控分子的立体化学特性,对手性联吡啶衍生物合成路线进行设计如Scheme1,对所合成的化合物进行抗肿瘤活性进行检测。
一、实验部分
(一)仪器与试剂。核磁共振仪(BRUKE-400);旋光仪(PerkinElmerModel341LC);高分辨质谱仪(JMS-800D);无水四氢呋喃、乙醚使用钠丝除水后重蒸,无水苯、甲苯、二氯甲烷使用钠丝除水,丙酮采用分子筛除水。
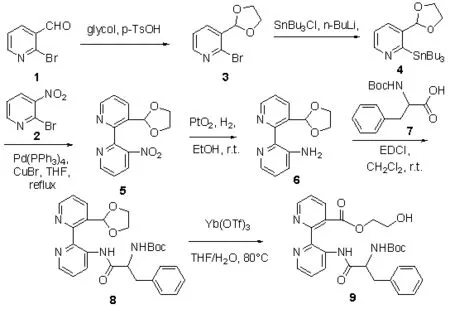
Scheme 1
化合物(3):2-溴-3-(1,3-乙二醇缩醛-2-烷基)吡啶按文献方法合成[4],其余试剂均为分析纯,其中无水四氢呋喃、乙醚使用钠丝除水后重蒸,无水苯、甲苯使用钠丝除水。
(二)化合物的合成。化合物(4):(三丁基锡)-3-(1,3-乙二醇缩醛-2-烷基)吡啶的合成。氮气保护下,将(3)(1.56g,0.68mmol,1.0equiv)溶于 3.0mL无水乙醚,在 -70℃下逐滴加入正丁基锂(0.96mL,1.6M),在-70℃下搅拌30min。用双针头管将溶于1mL无水乙醚的正三丁基氯化锡0.66mg(2.04mmol)转移至反应液中,在-70℃下搅拌0.5h。TLC检测反应结束后,加入水淬灭反应,用二氯甲烷萃取,饱和碳酸氢钠水洗,无水硫酸钠干燥有机相,减压蒸除溶剂,柱层析分离纯化(V(乙酸乙酯):V(石油醚)=1:40)蒸发溶剂得(4),无色油状物,收率 90;1HNMRδ:(dd,J=4.8Hz,2.0Hz,1H),7.73(dd,J=8.0Hz,1.4Hz,1H),7.10-7.20 (m,1H),5.7(s,1H),3.90-4.20(m,4H),1.50-1.70(m,6H),1.25-1.4(m,6H),1.15-1.25(m,6H),0.80-0.95(m,9H)
化合物(5):2-[3-(1,3-乙二醇缩醛-2-烷基)吡啶]-3-硝基吡啶的合成。在氮气的保护下,将(2)105mg(0.238 mmol)和(4)72.1mg(0.357mmol),溶于 7mL无水四氢呋喃中,加入 27.5mg(0.024mmol)三苯基磷钯,2.7mg(0.019 mmol)溴化亚铜,加热回流4.5h。TLC检测反应结束后,冷却反应液,减压蒸除去溶剂,柱层析分离纯化((乙酸乙酯):V(石油醚)=1:40)蒸发溶剂得(5),黄色油状物,收率85%;1H NMRδ:dd,J=4.6Hz,1.4Hz,1H),dd,J=4.8Hz,1.6Hz,1H)dd,J=8.2Hz,1.4Hz,1H),dd,J=7.8Hz,1.4Hz,1H),7.55 (dd,J=8.4 Hz,4.8Hz,1H),7.42(dd,J=8.0Hz,4.8Hz,1H),6.10(s,1H),3.80-3.95(m,4H);13CNMRδ:(154.0,153.1,152.0,149.4,145.8,135.6,132.6,132.4,132.1,132.0,131.9,128.6,128.5,123.6,123.5,100.5,65.1(2C);HRMS(ESI)calculated forC13H12N3O4[M+H]+:274.0822,found274.0828(-1.9ppm,-0.6mmu)
化合物(6):2-[3-(1,3-乙二醇缩醛 -2-烷基)吡啶]吡啶-3- 胺的合成。将(5)52.9mg(0.194mmol)溶于 8mL乙醇,加入 PtO2·3H2O5.45mg(0.019mmol),1atm 的氢气压力下催化氢化,反应进行4.0h,TLC检测反应结束。经过硅藻土过滤悬浮物,减压蒸除溶剂,柱层析分离纯化(V(乙酸乙酯):V(石油醚)=1:40)蒸发溶剂得(6),黄色半固体,收率 94%;1HNMRδ:(dd,J=4.8Hz,2.0Hz,1H),8.13(dd,J=8.0Hz,2.0Hz,1H),8.07-8.10(m,1H),7.60-7.70(m,1H),7.40-7.5(m,1H),7.34(dd,J=8.0Hz,4.8Hz,1H),4.60-4.8 (br,NH2),4.05-4.(m,2H),3.90-4.00 (m,2H);13C NMRδ:(156.5,148.5,142.4,141.2,138.6,133.6,132.1,132.0,131.9,128.6,128.5,124.0,123.8,122.9,100.3,65.5;HRMS(ESI)calcdlated forC13H14N3O2[M+H]+:244.1081,found 244.1089(-3.7ppm,-0.8mmu)
化合物 8(S)和 8(R)的合成通法。将(6)97.6mg(0.4 mmol)和(7)(60mmol)溶于 10mL 二氯甲烷中,加入 EDCI 191.7mg(0.6mmol),室温下搅拌,TLC 检测反应结束后,水洗(3×10mL),无水硫酸钠干燥有机相,过滤,减压蒸除溶剂,柱层析法分离纯化,得纯品。
8(S):无色蜡状固体,产率 80%,aD23=-24.4(c=2.5,CHCl3);1HNM(CDCl3):(br,1H),8.84(s,1H),8.68(s,1H),8.44(s,1H),8.3(d,J=2.8Hz,1H),7.40-7.50(m,1H),7.30-7.39(m,1H),6.4(s,1H),4.95(s,1H),4.30(s,1H),3.90-4.20(m,4H),1.60-1.9(m,2H),1.20-1.6(m,10H),0.93(d,J=6.4Hz,6H);13CNMR(CDCl3):(171.7,155.5,155.3,147.7,144.2,143.5,137.4,134.7,134.1,129.2,123.7,123.3,100.2,80.1,65.4,54.2,42.0,29.7,28.3,24.9,23.0,21.8;HRMS(ESI) calculated forC24H33N4O5[M+H]+:457.2445,found 457.2452(-1.4ppm,-0.7mmu)
8(R):白色固体,产率 47%,aD23=-40.8(c=4.0,CHCl3);1HNMR (CDCl3):(br,1H),8.83 (d,J=7.6Hz,1H),8.68(s,1H),8.40-8.48(m,1H),8.21(d,J=7.6Hz,1H),7.42(dd,J=7.6Hz,4.8Hz,1H),7.32(dd,J=8.6Hz,4.6Hz,1H),6.42(s,1H),4.95 (d,J=7.2Hz,1H),4.27 (d,J=2.8Hz,1H),3.90-4.15(m,4H),1.60-1.80 (m,2H),1.20-1.50 (m,10H),0.95(d,J=6.4 Hz,6H);13CNMR(CDCl3):(171.6,155.5,147.7,144.2,143.5,137.4,134.7,134.2,129.2,123.7,123.3,100.2,80.1,65.4,54.2,42.1,29.7,29.6,28.3,24.9,23.0,21.8;HRMS(ESI)calculated for C24H33N4O5[M+H]+:457.2445,found457.2454(-1.9ppm,-0.9mmu)
化合物 9(S)和 9(R)的合成通法。将(8)(50.0mg,0.1 mmol,1.0equiv)溶 于 THF-H2O(10mL,4:1v/v)Yb(OTf)3(76.15mg,0.12mol,1.1equiv) 室温下搅拌,TLC 检测反应结束后,二氯甲烷萃取反应液(3×10ml),无水硫酸钠干燥有机相,过滤,减压蒸除溶剂,快速柱层析法分离纯化,得纯品。
9(S):无色蜡状固体,产率 70%,aD23=-40.3(c=3.0,CHCl3);1HNMR(CDCl3):((br,1H),8.98(d,J=7.6Hz,1H),8.77 (s,1H),8.34 (dd,J=4.8Hz,1.2Hz,1H),8.02(d,J=7.2Hz,1H),7.30-7.60 (m,2H),4.60-5.10 (m,1H),4.10-4.60(m,3H),2.78 (d,J=13.6Hz,2H),2.20 (s,OH),0.90-1.99(m,18H);13CNM(CDCl3):(172.3,168.9,155.5,148.2,142.9,140.8,137.7,135.1,132.1,132.0,130.3,129.2,128.6,128.4,124.7,122.5,80.2,69.8,67.2,64.3,63.7,60.6,55.0,54.5,42.0,29.6,29.4,28.3,25.3,24.9,23.0,21.8;HRMS(ESI)calculated for C24H33N4O6[M+H]+:473.2395,found 473.2407(-2.7ppm,-1.2 mmu)
9(R):无色蜡状固体,产率 55%,aD23=+41.1(c=2.6,CHCl3);1HNMR(CDCl3):(br,1H),8.99(d,J=6.4Hz,1H),8.77(s,1H),8.35(dd,J=4.4Hz,1.6Hz,1H),8.02(d,J=6.8Hz,1H),7.30-7.50(m,2H),4.99(d,J=7.2Hz,1H),4.10-4.60(m,3H),3.80 (s,2H),2.22 (br,OH),0.90-1.95(m,18H);13CNMR(CDCl3):(172.3,169.0,155.5,155.3,148.1,142.9,140.8,137.6,135.1,130.4,129.2,124.7,122.5,80.2,67.3,60.7,60.4,54.5,42.1,29.7,28.3,24.9,23.1,22.6,21.8;HRM (ESI)calculatedforC24H33N4O6[M+H]+:473.2395,found473.2404(-1.9ppm,-0.9mmu)
(二)体外抗肿瘤活性测试。活性测试方法:将细胞接种于细胞培养板上,根据靶细胞不同选择每孔104~106个不同的细胞数。加入待测的化学药物,同时设相应的对照,置于37℃,5%CO2环境中培养,培养时间随测定内容及靶细胞不同而异。培养终止前4h加入5mg/mL的MTT溶液10~25ul/孔。终止培养,将所形成的结晶物用有机溶剂完全溶解后,在分光光度计上测定其再波长490nm或570nm处得A值。统计获得的实验数据,通过IC50计算软件,自动拟合生成曲线图活性因子的单位、效应细胞杀伤率等。
二、结果与讨论
(一)化合物的合成。化合物(8)的合成过程中,在还原产物(6)中引入不同光学纯的α-氨基酸作为手性源,反应中偶联剂DCC难以完全除尽,在1H NMR谱的高场有杂质峰存在,影响化合物结构鉴定。选用易于水洗除去的EDCI作为偶联剂,能够克服上述缺点。
(二)体外抗肿瘤活性。选择五种不同的肿瘤细胞株(HCT-8:人结肠癌细胞;Bel7402:人肝癌细胞;BGC-823:人胃癌细胞;A549:人肺腺癌细胞;A2780:人卵巢癌细胞)对目标化合物8和9的抗肿瘤活性进行了生物评价,见表1。

表1 目标化合物抗肿瘤活性筛选
评价数据结果表明,目标分子对所选用的五种肿瘤细胞株的增殖没有明显的抑制活性。随后,为了改变化合物的酯水分配系数,提高其跨膜能力,增强化合物与血管内皮生长因子的作用能力,对化合物8和9进行了化学修饰和结构改造。期待通过对化合物8和9结构的改造来调整化合物的酯水分配系数,提高分子的跨膜能力,有利于分子进入肿瘤细胞内部与血管内皮生长因子发生相互作用,提高化合物的抗肿瘤增殖的能力,期望发现与血管内皮生长因子具有高亲和力的新型先导化合物。
三、结论
本文设计并合成了一系列新型的含有手性中心的联吡啶类化合物,其结构经1H NMR,13C NMR和HRMS谱图确证,实验结果该方法对合成具有潜在生物活性的手性联吡啶化合物的有一定借鉴价值。MTT实验结果显示,目标化合物对 HCT-8、Bel7402、BGC-823、A549、A2780 细胞无显著抑制活性。
猜你喜欢
医学信息(2024年23期)2024-12-12 00:00:00
云南化工(2021年10期)2021-12-21 07:33:28
化工学报(2020年4期)2020-05-28 09:25:24
今日农业(2019年11期)2019-08-13 00:49:02
中成药(2017年10期)2017-11-16 00:50:15
中成药(2017年4期)2017-05-17 06:09:46
中国医药生物技术(2015年4期)2015-12-26 08:26:36
中国现代医学杂志(2015年26期)2015-12-23 11:04:22
西南国防医药(2015年11期)2015-02-28 19:38:46
疑难病杂志(2014年12期)2014-04-16 05:19:29
