
甲钴胺的环境友好生产工艺过程研究
2018-08-14 08:34:34
分析仪器 2018年4期
(1.山东大学药学院,济南 250012; 2.石药集团中诺药业(石家庄)有限公司,石家庄 050051)
甲钴胺属于辅酶型的一种维生素,甲钴胺甲基化的官能团能够参与到体内甲基转移作用中,从而对体内神经组织核酸、蛋白质和脂肪的新陈代谢等都具有良好促进作用。在人体代谢中,甲钴胺和腺苷钴胺能够直接参与体内代谢,对疾病起到直接治疗作用。从甲钴胺的结构形式来说,甲基VB12要比普通氰钴VB12疗效更好,因此可以说甲基VB12未来将是氰钴VB12的一个更新换代产品。国内外学者在对甲钴胺临床作用研究中发现甲钴胺对于糖尿病的周围神经病变拥有良好治疗作用,能够改善糖尿病周围神经的形态,让神经损害得以修复。此外,甲钴胺还在三叉神经痛、面神经炎治疗等领域有良好治疗作用。
在这种情形下,开展甲钴胺原料药工艺技术研究开发与生产,生产出成本低、质量高、对环境污染小的产品,满足临床用药需求,具有重大现实意义。
1 材料与方法
1.1 主要仪器与原料
甲钴胺制备与质量控制的材料见表1。

表1 甲钴胺制备与质量控制的材料
1.2 制备路线
氰钴胺→还原→还原态氰钴胺→甲基化→ 甲基氰钴胺溶液→树脂吸附→洗脱液→结晶→干燥→成品→包装。
1.3 质量控制
1.3.1 鉴别
(1)避光操作,分别取本品和对照品各约5 mg,置100 mL容量瓶中,加水溶解,定容至刻度,摇匀备用。照紫外-可见分光光度计标准操作规程,进行波长扫描检测,供试品溶液在220~550 nm的波长范围内的吸收光谱应与对照品溶液的一致。(在266 nm、342 nm、525 nm处有最大吸收)。

(3)在含量测定项下记录的色谱图中,供试品溶液主峰的保留时间应与甲钴胺对照品溶液主峰的保留时间一致。
(4)取本品按照红外光谱测定法进行红外光谱测定后,将本品的红外光吸收图谱应与对照的图谱(光谱集732图)一致。
1.3.2 水分测定
精密称定本品约0.1g,按照卡尔费休水分滴定仪标准操作规程测定。同法测定对照品。
偏差:同样方法同时测定3个平行样,3个数据的RSD应≤5.0%,取平均值报告。
1.3.3 含量测定
流动相的配制:
称取磷酸二氢钾4.1 g,置1000 mL容量瓶中,加纯化水溶解并稀释至刻度,摇匀,用磷酸调节pH值为4.5后,用0.45 μm的水系微孔滤膜过滤,即得0.03 mol/L磷酸二氢钾溶液。将乙腈与0.03 mol/L磷酸二氢钾溶液按16∶84的比例混合均匀,放入超声波水浴中脱气10分钟,即得。
供试品溶液的配制:
称取本品25±2.5 mg于称量瓶内,将称量瓶放入漏斗中,将样品经漏斗倒入50 mL容量瓶中,用流动相冲洗称量瓶及漏斗,洗液并入容量瓶中,加流动相稀释至刻度摇匀,再精密量取5 mL,置于50 mL容量瓶中,加流动相稀释至刻度摇匀,即为供试品溶液。供试品溶液使用前经油系微孔滤膜(0.45 μm)过滤。
对照品溶液的配制:
取甲钴胺对照品同供试品溶液配制方法,配制对照品溶液。
色谱条件:
色谱柱:C18柱;检测波长:342 nm;柱温:40 ℃。
流速:调节流速使甲钴胺峰的保留时间约为12分钟(11.5分钟至12.5分钟)。
测试过程:
照液相色谱仪标准操作规程依法操作。取对照品溶液与供试品溶液各20 μl注入液相色谱仪,记录色谱图。按外标法以峰面积计算,即得。
系统适用性:
系统表现:取甲钴胺对照品25±2.5 mg,置50 mL容量瓶中,加水溶解并稀释至刻度,摇匀,在自然光下放置5~10分钟,取20 μL注入液相色谱仪,调节流速使甲钴胺峰的保留时间约为12分钟,记录色谱图,甲钴胺峰与羟钴胺峰(相对保留时间约为0.2)的分离度应大于20,甲钴胺峰与相邻杂质峰之间的分离度应>1.5。
系统重现性:取对照品溶液20μL注入液相色谱仪,重复进样5针,所得5张图谱中主峰面积的RSD≤2.0%。
偏差:取对照品K值的平均值计算,对于同一种方法要同时测定两个平行样,RD应≤0.5%。

K对:对照溶液主峰面积/所称对照品的重量(g);
K样:供试品溶液主峰面积/所称供试品的重量(g);
C:对照品百分含量;
W对:对照品水分;
W样:供试品水分;
A:对照溶液(供试品溶液)主峰面积;
G:所称对照品(供试品)的重量(g)。
1.3.4 有关物质
流动相的配制:
称取磷酸二氢钾4.1 g,置1000 mL容量瓶中,加纯化水溶解并稀释至刻度,摇匀,用磷酸调节pH值为4.5后,用0.45 μm的水系微孔滤膜过滤,即得0.03 mol/L磷酸二氢钾溶液。将乙腈与0.03 mol/L磷酸二氢钾溶液按16∶84的比例混合均匀,放入超声波水浴中脱气10分钟,即得。
供试品溶液的配制:
取本品25±2.5 mg于称量瓶内,将称量瓶放入漏斗中,将样品经漏斗倒入50 mL容量瓶中,用流动相冲洗称量瓶及漏斗,洗液并入容量瓶中。加流动相稀释至刻度,摇匀,作为供试品溶液。使用前经0.45 μm的油系微孔滤膜过滤。
对照液配制:
精密量取供试品溶液0.5 mL,置50 mL容量瓶中,加流动相稀释至刻度摇匀,作为对照液。使用前经0.45 μm的油系微孔滤膜过滤。
色谱条件:
色谱柱:C18柱;检测波长:342 nm;柱温:40 ℃。
流速:调节流速使甲钴胺峰的保留时间约为12分钟(11.5分钟至12.5分钟)
测试过程:
照液相色谱仪标准操作规程依法操作。取供试品溶液与对照溶液各20 μL,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的3倍。记录供试品溶液的色谱图中最大单个杂质峰面积、杂质峰面积之和及对照液主峰面积。
偏差:同一方法测定两个平行样,RD应≤5.0%,取两个结果的算术平均值报告。
计算公式:


2 甲钴胺制备工艺研究
2.1 还原反应的催化剂及限制试剂的选取
根据专利和文献报道[1-3],国际上制备甲钴胺所采用的还原剂为硼氢化钠。但硼氢化钠是一种强还原剂,直接把维生素B12的中心三价钴离子还原成一价态、能把β位上氰根还原掉,也使α位上的苯并咪唑键断裂,因此在产物中容易生成无咪唑基的钴啉醇酰胺类化合物,反应不易控制。又因该反应以铁盐做催化剂,对环境造成污染。基于多年甲钴胺制备的经验与文献资料[4]的查阅,选择了两种还原催化剂Por-A和Por-B、一种限制试剂QAS。该两种催化剂都可迅速催化硼氢化钠水解,而且都可以经过再生后重复利用,不对环境造成固废污染。因在反应中加入了限制试剂,在还原过程中不会把氰钴胺分子中的三价钴还原成一价钴。我们对以上两种催化剂进行了对比试验,实验数据见表2,由表2数据做出的甲钴胺的转化率随时间的变化曲线见图1。

表2 不同催化剂的转化率 %
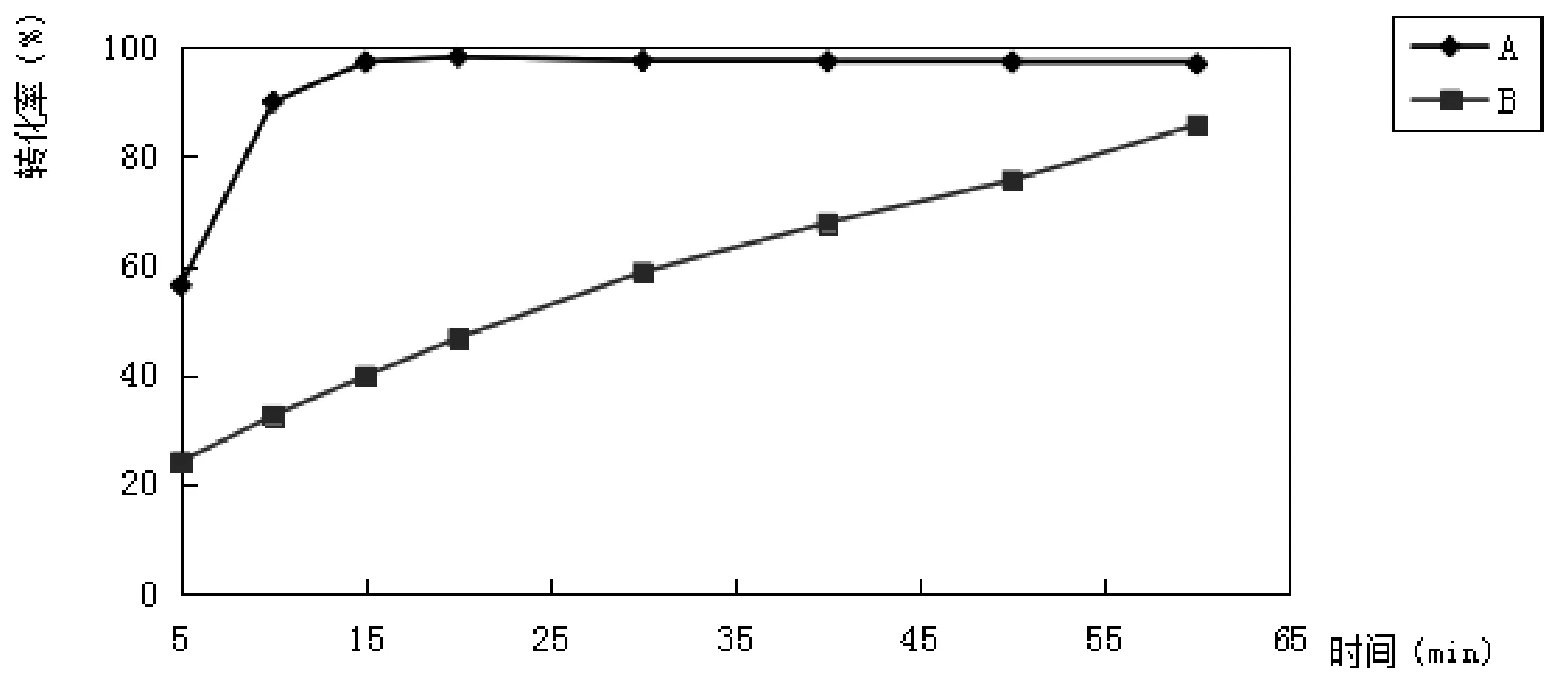
图1 不同还原反应催化剂的转化率
由表2及图1可以看出,催化剂Por-A的催化能力优于催化剂Por-B,因此选择催化剂Por-A作为该工艺的还原反应催化剂,在30±2 ℃的温度下,其最佳还原时间为20分钟。
2.2 选择新型甲基化试剂的原因和应用效果
目前甲钴胺的生产工艺中普遍采用的甲基化试剂是碘甲烷或三甲基碘化亚砜,碘甲烷是一种良好的亲核试剂,但是碘甲烷的沸点只有42 ℃,挥发性极强,而且毒性也很大,因此这种工艺用料对操作人员的健康与生命极具威胁。尤其它不溶于水还要用甲醇或者乙醇作溶剂,为此在精制前还通过蒸发的方式除去溶剂,这一过程又会使甲钴胺受热破坏,从而降低收率并使产品质量下降。虽然三甲基碘化亚砜无毒又溶于水,但它甲基化后会生成具有恶臭的硫醇类物质随着尾气逸出到空气中,这又造成了大气污染。为此课题组根据文献[5,6]及生产经验,选择了一种全新的甲基化试剂M-S,该种试剂是一种固体、无毒无味、易溶于水,且甲基化后不产生异味物质。经试验证明其甲基化效果良好,可以使反应转化率达99%以上,新型甲基化试剂与三甲基碘化亚砜的对比效果见表3。
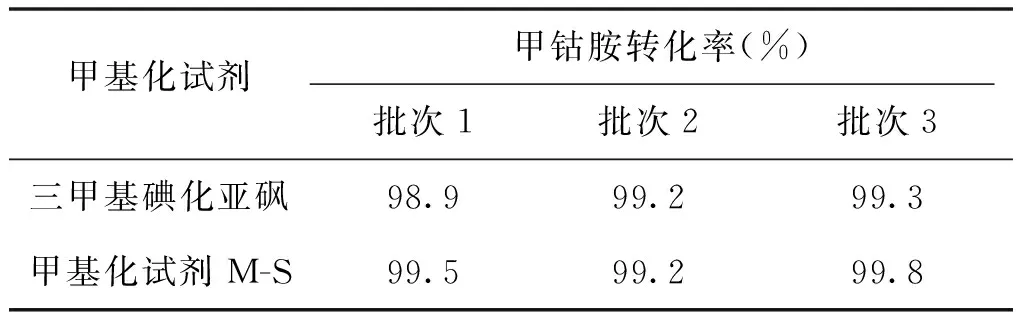
表3 新型甲基化试剂与三甲基碘化亚砜的对比效果
根据表3的数据可知新型甲基化试剂M-S在同样条件下能够达到三甲基碘化亚砜的效果,同时减少了废气的排放。
2.3 反应温度、原料浓度及限制试剂加量反应时间对甲钴胺转化率的影响
在确定催化剂Por-A、限制试剂QAS和甲基化试剂M-S后,我们以原有的工艺条件为基础对其工艺条件进行优化。经分析[7]我们认为反应温度、氰钴胺投料浓度及限制试剂的加量将是影响甲钴胺转化率的主要因素。如果反应温度过高会导致反应过于剧烈,使VB12分子降解,影响产品质量和收率;如果反应温度过低,导致还原及甲基化反应不完全,同样影响产品的质量与收率。在该反应中,还原剂水解速度的限制试剂加量也同样是一个重要参数,因此我们以反应温度、原料浓度及限制试剂的加量为影响因素,以正交试验法来获得最佳反应条件(以甲钴胺的转化率为目标函数)。试验方案的因素水平见表4。

表4 因素水平表
按照正交试验方案进行试验,试验结果见表5,根据试验结果对数据进行方差分析以确定最佳反应条件,方差分析表见表6。
表6数据可以看出: FA=57.02, FB=22.50大于F0.90(2,2)=9.0,也大于F0.95(2,2)=19.0,因此反应温度与甲基化时间两因子在显著性水平0.10与0.05上都是显著的。而FC=2.47小于F0.9(2,2)=9.0,故物料浓度不显著,可忽略。
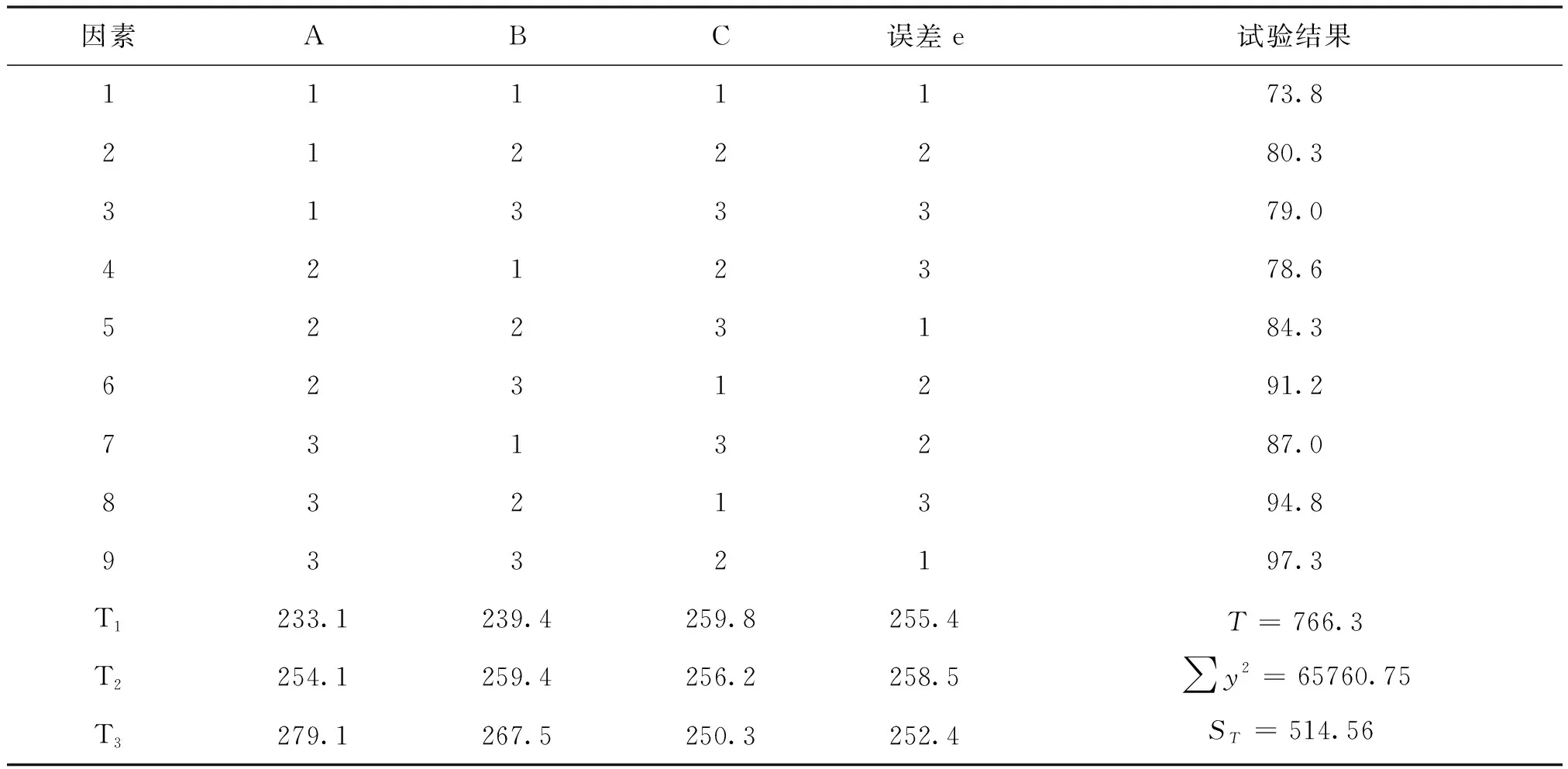
表5 反应温度、原料浓度及甲基化时间的影响

表6 方差分析表
表5、表6的计算结果显示: A3水平为最佳温度条件,即40±2 ℃。B3水平为最佳甲基化反应时间。对于不显著物料浓度项,参考原有氰钴胺的投料浓度,以2%为本研究的投料浓度。
正交试验完成后,以选取的工艺条件进行验证,验证数据见表7。

表7 三批甲钴胺数据
从表7数据可以看出:以正交实验确定的工艺条件可以实现甲钴胺的高转化率。把该条件作为甲钴胺的合成条件。
2.4 甲钴胺精制介质的选取
现代医药技术要求原料药的色普纯度较高[8],对于维生素B12品种来说国际上一般要求其相关物质以高效液相色谱法检测不得大于2.0%。我们为了使得甲钴胺原料药在国际上更具有竞争力,将目标定为相关物质控制在1.0%以内。为此课题组与介质生产厂家合作生产了5种精制介质:JZ-1、JZ-2、JZ-3、JZ-4、JZ-5。对5种介质的精制性能进行了试验研究。试验结果如表8。

表8 精制介质的分离效果
注:表中分离效果是指在精制介质上杂质色带与主色带的分离程度。
从表8看出,精制介质ZJ-3分离相关物质的能力最好。
3 质量研究
按照国内外相关法律法规对新工艺所生产的产品进行了稳定性考察[9]与质量研究。结果表明:新工艺生产的甲钴胺产品的各项质量指标均符合内定的质量指标要求。
3.1 各国药典标准及产品内控标准[10](表9~表12)
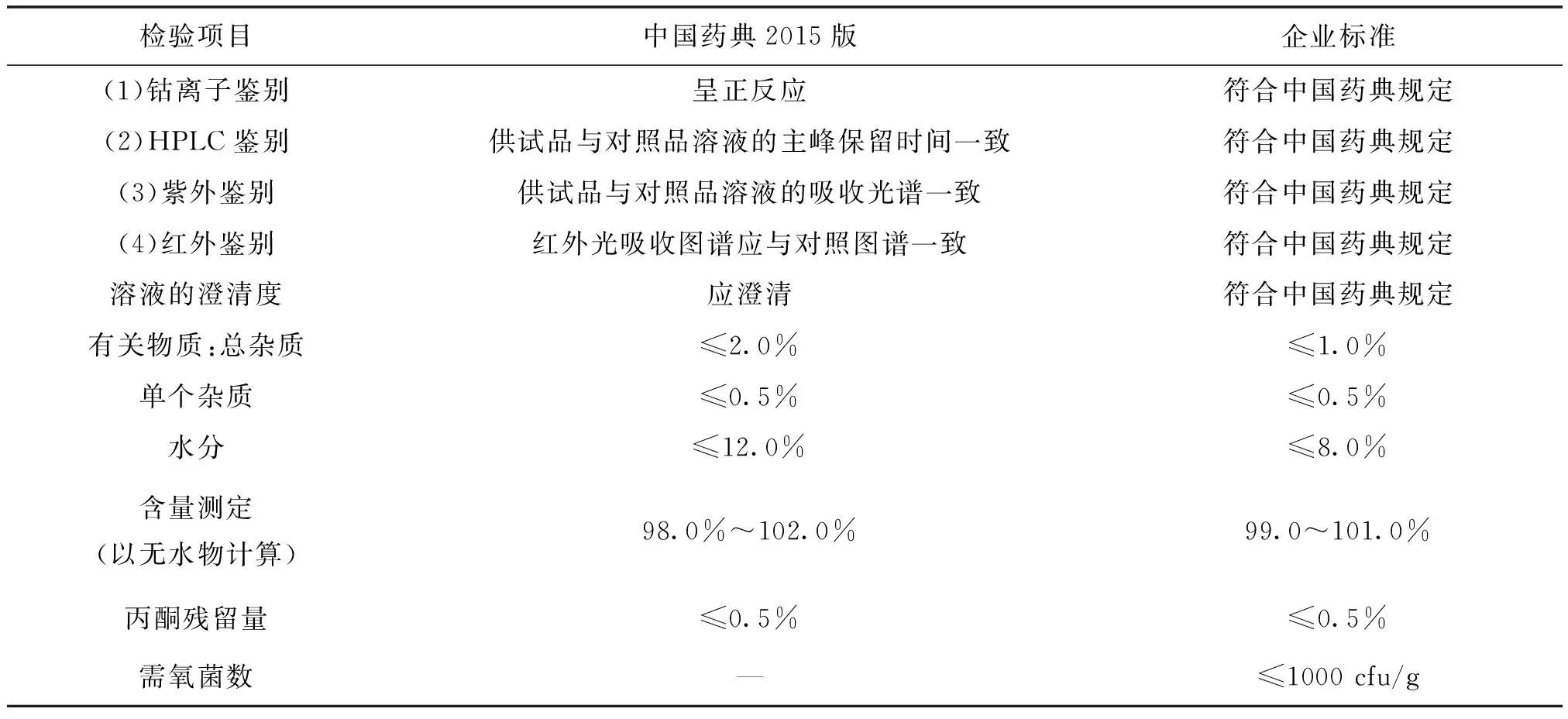
表9 国内市场标准(CP)

表10 日本市场标准(JP)

表11 印度等市场标准(JP)

表12 美国市场标准(USP)
3.2 结构简单确认
把以本课题研究确定的制备工艺制得的甲钴胺与甲钴胺标准品进行红外光谱确认,以溴化钾压片法制备测试样品。
结论:甲钴胺样品的红外图谱与甲钴胺标准品的红外图谱基本一致。故从其红外光谱上可以证明以新工艺制备出的产品为甲钴胺。结果表明:
(1)新工艺生产的甲钴胺产品的各项质量指标均符合内定的质量指标要求;
(2)与工艺开发前的产品对比研究表明,新工艺生产的甲钴胺纯度高,质量明显优于工艺开发前的产品;
(3)在已完成的加速稳定性考察过程中,产品重点质量指标均符合各相关药典规定。
4 结果与讨论
本工艺研究取得的主要成果与结论简述如下:
(1) 提高了还原反应转化率:本课题确定了新型催化剂及还原反应的工艺条件,该研究采用两种不同的还原反应催化剂,经过大量的对比试验,选出了适合甲钴胺合成反应中还原工序适宜的催化剂及相应的工艺条件,有效控制了还原剂的还原速度,使得氰钴胺能够快速生成二价态的B12。
(2) 满足了清洁生产的要求:由于在还原反应阶段使用了新型催化剂,由于该催化剂能够重复利用,在提倡可持续发展的今天,减少了原料药生产中三废的产生,有效的满足了清洁生产的要求。
(3) 提高产品品质,降低三废:本课题选取了新的甲基化试剂,该甲基化试剂具有溶于水、甲基化能力强、不产生异味物质等优点,不仅可以生产出满足内控质量标准的产品,同时对员工身体健康、环境保护做出了贡献。
总之,以本课题研发的生产工艺生产的甲钴胺原料药具有收率高、质量好、成本低、清洁污染少等优点。在满足患者临床用药安全性、提高公司市场竞争力方面具有重大意义。
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12 02:07:48
中学生数理化·自主招生(2022年4期)2022-05-09 22:00:23
中学化学(2019年4期)2019-08-06 13:59:37
中国调味品(2017年2期)2017-03-20 16:18:13
Cancer Biology & Medicine(2016年4期)2017-01-13 01:54:45
中学化学(2015年2期)2015-06-05 07:18:13
现代检验医学杂志(2015年2期)2015-02-06 02:00:48
沈阳医学院学报(2014年4期)2014-12-27 13:44:30
遗传(2014年3期)2014-02-28 20:58:49
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28 12:22:07
