
表面活性氧物种对A-H(A=C,O,N)键活化机理的研究进展
2018-08-08 10:33王贵昌
信阳师范学院学报(自然科学版) 2018年2期
王贵昌
(南开大学 化学学院,天津 300071)
0 引言
在多相催化反应中,A-H (A=O,N,C)的分解是许多重要多相催化反应的初始步骤,如水煤气变换反应及蒸汽重整反应过程中H2O分子O-H的断裂,天然气合成中甲烷分子C-H键的活化,甲醇,乙醇,甲酸分解产生H2能源中O-H键的活化,以及NH3分子中N-H键的活化等.实验表明,在IB族金属如Cu、Ag、Au上,这些键的活化非常困难,尤其在Ag、Au表面.有趣的是,当这些催化剂表面存在活性氧物种如O*、OH*时,A-H键的活化程度会有明显的提高[1-16].最初表面科学家MADIX课题组发现Cu/Ag/Au表面吸附态氧物种作为布朗斯特碱可以促进如H2O(或 CH3OH、C2H5OH、HCOOH)分子中O-H中强酸性质子H的活化,或乙炔/丙烯中C-H键的活化[6-12].后来人们发现同样的促进作用也可以发生在非贵金属如Pt[13]、Pd[14]催化的有机酸的分解反应,并且近期的研究表明表面氧原子对于Pd、Pt、Rh催化的甲烷部分氧化反应同样具有促进作用[15,16].为探讨活性氧物种对于A-H键活化促进作用的内在本质,理论科学工作者在该方面进行了大量而深入的研究工作,其中包括经验方法以及基于密度泛函理论的第一性原理的理论计算[12,17-39].通过系统地理论研究,人们发现一个普遍的规律:A-H的促进作用与氧原子在金属表面的吸附强度有关,吸附越强,促进作用越弱.同时还发现活性氧物种的碱性强弱与其吸附强度有很好的关联,即吸附越强,其碱性越弱 (见图1).
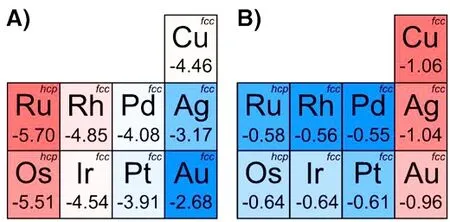
图1 A) OH*在过渡金属表面的结合能(BE,eV) 和 B)电荷(|e|) [12]Fig. 1 A) Binding energies (BE, eV) and B) charges(|e|)for OH* on transition metal surfaces[12]
该基本规律对于如何调控A-H键的活化程度提供了一种新的思路.本文概述了近些年来该领域的理论研究成果(主要是基于密度泛函理论的第一性原理的研究),期望对实验中有关A-H键活化的调控提供一定的理论指导作用.
1 O对A-H 键活化的影响
有关O物种对于A-H键的影响,最初的理论工作来源于SHUSTOROVICH等人的基于经验的键级守恒方法(BOC-MP 或UBI-QEP)的研究[17-20].他们通过比较X-H直接分解(X-H(ad)X(ad)+H(ad))与氧参与的间接分解(X-H(ad)+O(ad)X(ad)+OH(ad)的反应活化能发现,吸附态氧原子可以促进Cu/Ag表面催化的X-H键的反应活性,而对于W/Ni表面而言则为抑制作用,即氧原子的速进作用与金属的性质有关,且氧原子的促进作用与形成的强吸附-OH基团有关[17].随后,Au等人采用金属簇模型结合DFT-UBI-QEP方法对氧原子影响的甲烷C-H键活化规律进行了系统的理论计算研究[18,19].计算结果表明,当氧原子结合在金属顶位时可以促进甲烷的脱氢(CH4,sCs+4Hs),而当氧原子结合在穴位时,只对Pt/Cu/Ag/Au金属具有促进作用,对其他金属如Ru/Rh/Os/Ir/Pd则表现为抑制作用(见表1).
表1 吸附态氧存在与不存在情况下解离能的差别(in eV)[19]Tab. 1 Difference (in eV) between the dissociation energieswith and without the involvement of chemisorbed oxygens[19]
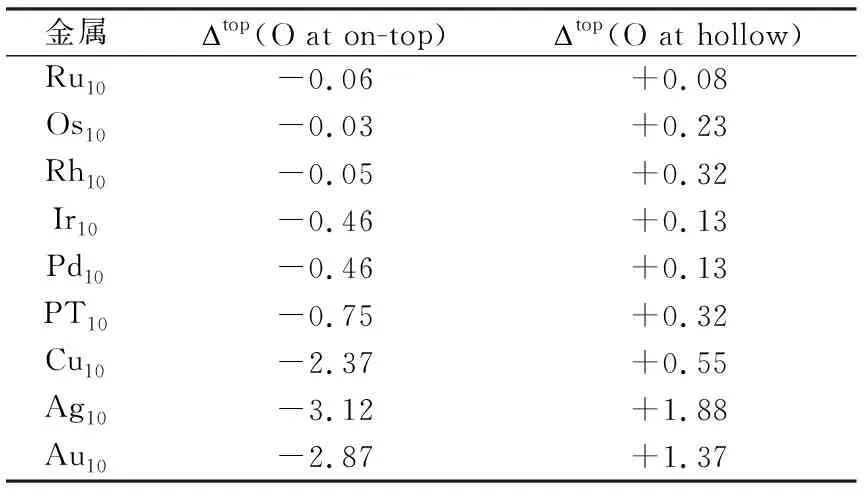
表1 吸附态氧存在与不存在情况下解离能的差别(in eV)[19]Tab. 1 Difference (in eV) between the dissociation energieswith and without the involvement of chemisorbed oxygens[19]
鉴于经验方法UBI-QEP的不足,人们更多地采用基于密度泛函理论(DFT)的第一性原理来研究活性氧物种对X-H键活性的影响.我们曾采用DFT方法对一系列过渡金属(包括VIII族金属和IB金属)催化的H2O分子中O-H键的活化以及吸附态氧原子的作用进行了系统地理论研究[21],结果表明氧原子的促进作用与氧原子在金属表面的吸附强度有关:氧原子在表面吸附越强,对O-H键活化的促进作用越弱(见图2).H2O+OOH+OH反应中氧原子的作用包含两个方面: 吸附态氧原子一方面与金属表面结合,另一方面与H2O作用.显然与金属作用越强(即吸附热越大),则与H2O的作用就越弱,即对O-H键的活化程度越弱(有时甚至表现为抑制O-H键分解的作用,如Mo, Re).类似地,NEUROCK等人[12]也得到了相同的规律,并且发现氧原子的吸附强度与其碱性强弱有关:若吸附越强,则其碱性越弱,相应地促进作用就越弱(因A-H中的H表现为酸性).

图2 水在清洁及氧修饰表面分解活化能差值与氧原子吸附热之间的关系[21]Fig. 2 Relationship of reaction barrier difference forwater dissociation on cleanand oxygen preadsorbed metal surfaceand the adsorption energy of atomic oxygen[21]
对于吸附态氧原子对A-H键活化的影响,除了与金属催化剂的特性有关外,还与A-H键的特性有关.对于甲烷分子中C-H键的活化,我们的理论计算结果表明氧原子在IB族金属如Cu/Ag/Au上表现为促进作用,而在其他过渡金属上则表现为抑制作用[25](见图3a),这与H2O的分解情况是不一样的[21],因为氧原子对于H2O的分解几乎都有促进作用(除Mo等极少数金属外[23]).同样地,NEUROCK等人的工作也得到了相同的规律[12].进一步地,系统的理论计算表明吸附态氧原子对O-H键的活化程度最大(即促进作用越大),N-H 次之,C-H键的活化程度最弱[26](见图3b).该顺序可能与X-H 键的极性顺序以及氢键形成的难易程度有关,也可能与质子H的酸性强弱有关,如甲醇中H的pKa=17, 而甲烷中H的pKa=56.
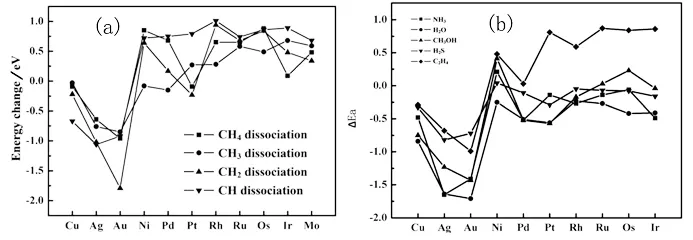
图3 (a)清洁与氧修饰金属表面甲烷分解活化能的差值随金属性质变化[25],(b)一系列A-H键分解活化能差值(△Ea)与O-H, C-H 和N-H键的关系[26]Fig. 3 (a) Activation barrier difference with and without oxygenfor different methane dissociation steps on various metal surfaces[25] ,(b) andbarrier difference (△Ea) with and withoutoxygen atom for the O-H, C-H and N-H bond[26] (b)
对于Pt(111)与Rh(111)催化的NH3分子中N-H键的活化,理论计算结果表明:吸附态氧原子对Pt(111)催化的NH3的脱氢具有促进作用,对NH2/NH的进一步脱氢基本无影响;Rh(111),表面存在的氧原子则对NH2/NH的脱氢表现为抑制作用[35].其原因在于NH2或 NH体系,过渡态中NH2(或NH)与O共用金属原子(因NH2或NH与O均倾向于结合在桥位或穴位),存在斥力,导致活化能升高;同时由于O原子在Rh(111)上的吸附强度强于Pt(111),因而其斥力作用更强,使N-H键分解的活化能进一步升高.对于同一金属,当其晶面不同时,氧物种对A-H的活化能力也不尽相同.对于Pt(100)面,NH2基团的分解活化能在氧原子存在下则变低了[36,37](见图4).原因在于由于(100)面比较开阔,过渡态时不需要共用金属原子,则排斥力降低,有利于过渡态的稳定.
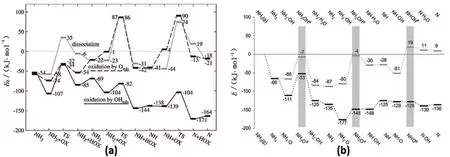
图4 Pt(111) (a)和 Pt(100) (b) 面上O和OH 对于NH3氧化脱氢的势能图[36,37]Fig. 4 Reaction pathway for the oxidative dehydrogenation of ammonia over Pt(111) (a) and Pt(100) (b) assisted by O and OH[36,37]
我们也曾对Cu(111), Cu(100), Cu(110) 催化的H2O分解过程进行了理论计算研究[27,28],发现在清洁表面的活性顺序为 Cu(110) > Cu(100) > Cu(111), 而在氧原子存在下,虽然其活性普遍升高,但其活性顺序变为Cu(110) > Cu(111) > Cu(100), 即Cu(100)上氧原子的促进作用最弱,这与其吸附氧原子能力最强有关[27].研究结果还显示单原子铜催化剂上氧原子对H2O分解的促进作用最强,一方面是由于其结合氧原子的能力比较弱,另一方面是由于氢键的形成[28].此外,一个有趣的现象是“Cu-O”双活性位酸碱对的作用强弱与H2O的分解活性近似线性相关,即“Cu-O”双活性位的协同作用促进O-H键的分解[28].
上面讨论的对象针对的是诸如H2O、CH4等小分子中O-H、C-H的直接解离,对于较为复杂的分子如CH3OH、C2H5OH等,还包含有C-H、C-H等,其作用机理将更为复杂.XU等人[32]通过理论计算研究了Au(111) 表面上O原子对甲醇耦合反应生成乙醚等的反应过程,发现O原子除了能促进O-H的分解生成CH3O外,对CH3O物种中-H的消除生成甲醛过程同样具有促进作用(活化能由0.64 eV 降低为 0.49 eV),但其促进作用弱于O-H的分解(反应CH3OHCH3O+O的活化能从清洁表面的1.58 eV 减小为氧存在下的0.41 eV),同样也说明O-H的活化程度强于C-H,与前面的结论相一致[26].
2 OH 对A-H 键活化的影响
在碱性溶液中,或在H2O存在条件下,催化剂表面可能存在OH物种,其对A-H的活化同样会有比较大的影响.NEUROCK 等[12]研究了OH对CH3OH分子中O-H分解性能的影响,发现OH的促进作用往往强于O原子,并且吸附越弱,促进作用越强(见图5a).同理,对于CH4分子中C-H键的活化,有类似的作用规律,只是促进作用较O-H弱些(见图5b).进一步比较还发现在VIII过渡金属上,OH基团对O-H分解的活化能小于O原子参与时的活化能,但在VI族金属上则相反,即O的促进作用强于OH, 但它们的存在均较直接解离有利.OH的促进作用强于O的可能原因在于OH的吸附强度弱于O,因而碱性强.对于NH3分子中N-H键的活化,O与OH的影响也曾有过系统的理论研究[36,37](见图4).在Pt(111)表面,O原子只能促进NH3分子的分解,而OH可以促进所以NHx物种的分解,而在Pt(100)表面,O原子的促进要强于OH.
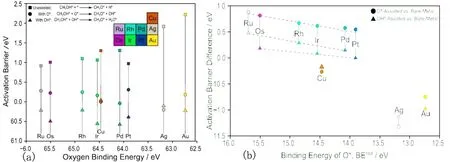
图5 甲醇分子中O-H键的直接解离,O参与以及OH参与的间接解离的活化能(a)甲烷分子O与OH参与解离活化能与直接解离活化能差值与氧原子吸附热之间的关系(b)[12]Fig. 5 Activation barrier for the O-Hactivationofmethanol through direct, O-assisted and OH-assistedmechanisms (a) and difference between O-assisted and direct CH4 activation barriers and difference between OH-assisted and direct CH4 activation compared to the bindingenergy of O(b)[12]
对于较为复杂的有机分子,如5-羟甲基-2-呋喃的绝热氧化反应,当用Pd做催化剂时,理论计算结果表明OH对C-H以及O-H的活化具有强的促进作用,且Pd(111)的促进作用强于Pd(100)面,这与OH在Pd(111)、Pd(100)面上的吸附强度顺序相一致[29](图6),即OH在Pt(111)表面的吸附较(100)面弱,因而其促进O-H/C-H分解的作用就强.
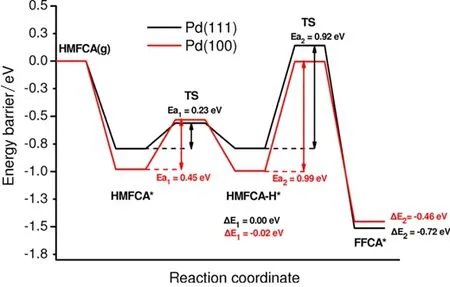
图6 Pd(111)和Pd(100) 表面5-羟甲基-2-呋喃羧酸氧化为5-甲酰基-2-呋喃甲酸势能图[29]Fig. 6 Potential energy profiles for the oxidation step from5-hydroxymethyl-2-furancarboxylic acid (HMFCA) toward5-formyl-2-furancarboxylic acid (FFCA) on Pd(111) and Pd(100)[29]
3 晶格氧对A-H键活化的影响
除了金属表面的吸附态氧原子外,金属氧化物中的晶格氧原子(O2-)同样会影响X-H的活性.HU等人[30]研究了CH4在不同氧化物如 PdO(001)、PdO(110)、PdO(100)上的分解性能,发现CH4的分解活性与晶格氧结合质子H的能力有关,即晶格氧的碱性越强,结合质子的能力就越强,则分解CH4分子的活性就越高.这是由于CH4在金属氧化物的分解过程为一“异裂”过程,其中金属Pd与CH3结合,O与H结合,因此[Pd-O]酸碱对的强弱决定了CH4分子的活化程度.类似地,近期的第一性原理计算结果表明[31],甲烷分子在Cu(111)表面分解活化能很高(169. 8 kJ/mol,而在CuO表面上由于晶格氧的存在,它可以与表面配位不饱和Cu组成Cu-O酸碱对协同活化甲烷分子中的C-H键,从而使分解活化能大幅度降低,其中在不稳定CuO(010)表面上的活化能为60.5 kJ/mol,在最稳定的CuO(111)表面上C-H分解活化能为76.6 kJ/mol.如此低的C-H分解活化能是由于稳定的过渡态(TS)、降低的应变能以及热力学上稳定的共吸附产物等原因所引起的.同样的解释也可用于金属表面,即氧原子的存在使C-H键的活化由纯金属的单活性中心的三位活化机理(CH3-Cu-H)变为双中心的四位活化机理(CH3-(Cu)-H-(O)),其中Cu-CH3和 O-H 之间的偶极相互作用可以稳定过渡态,同时较短的过渡态键长也降低了甲烷分子的应变能,从而较单中心更为有利(见图7).
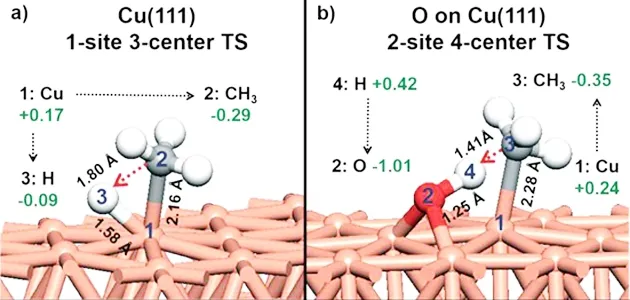
图7 甲烷在Cu(111) 和O修饰的Cu(111)表面不同解离机理的过渡态:a) Cu(111)表面的Cu单位三中心机理,b)O修饰的Cu(111)表面Cu-O双位四中心机理[31]Fig. 7 Transition states (TSs) for dissociation of methane on Cu(111)and oxygen pre-covered Cu(111) surfaces via different mechanisms:a) Cu single-site-three-centered mechanism on Cu(111) surface,b) Cu-O two-site-four-centered mechanism onchemisorbed oxygen containing Cu(111) surface[31]
4 超氧或过氧物种等对A-H键的活化
在氧气气氛下,吸附态的氧分子通过与催化剂表面的电子传递会发生一系列的结构和性能的变化[34]:O2(g)→O2(ads)→O2-→O22-→2O2-.由于这些氧物种的电子结构不同,其氧化脱氢的能力也各不相同.丙烯环氧化反应生成环氧丙烷(PO)是一重要的化工反应类型,但其甲基上的H易发生氧化脱氢反应(AHS)而生成丙烯醛甚至CO2,降低PO的选择性.一般而言IB族金属如Cu/Ag/Au是优良催化剂.在氧化反应气氛中,其表面存在的氧物种包括吸附态氧原子(O*或O-),同时也包含未解离的吸附态氧分子(O2*或过氧离子O22-),它们均能参与AHS反应.计算结果[39]显示吸附态氧参与的AHS反应的活性高于过氧离子的反应活性,并且反应AHS反应活性与质子H在相应氧物种上的结合能近似线性相关,即H结合越强,对于C-H的分解促进作用也越强(见图8).
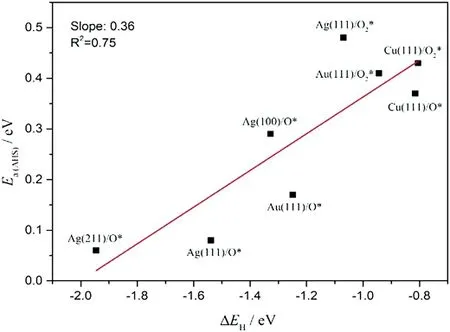
图8 吸附氧原子上H 亲和力EH与AHS反应活化能Ea(AHS)之间的线性关系[39]Fig. 8 The linear relationship between the hydrogen affinityof the adsorbed oxygen species EH andthe activation barrier for the AHSreaction Ea(AHS)[39]
我们最近的有关丙烯在Cu2O表面的选择性氧化的计算结果也表明,脱氢反应(AHS)的活性顺序为吸附态氧>吸附态氧分子>晶格氧, 即晶格氧的脱氢活性最差,原因在于其与Cu+结合太强的缘故. PALMER 等人[34]研究CH4分子在La2O3分解的过程中发现了类似的研究规律,即吸附态氧原子较吸附态氧分子具有更强的活化C-H 键的能力,并且发现晶格氧(O2-)的活化能力最弱.同样地,不同氧物种的夺氢能力与其结合质子氢的结合强度是相一致的,即O-> O22-> O2-.
5 结语与展望
综述活性氧物种对于脱氢能力大小的理论研究,可得到如下主要结论:(1)一般地,氧原子在金属催化剂表面结合越强(IB族金属较VIII金属强),其碱性就越弱,相应的促进作用就越弱;(2)OH较O的吸附强度弱,其促进作用就较O强些;(3)对于金属表面氧化物,一般地,脱氢活性顺序为吸附态氧原子>吸附态氧分子>晶格氧,与其结合质子H的能力相一致.(4)H的酸性强弱:O对O-H键分解促进作用最为明显,N-H活化次之,C-H 键最弱,这与其酸性强弱相一致.
由上述观点,可以得出一个优良的活化A-H键的催化剂应具有“M-O”双位活性中心,其协同作用促进A-H键的活化.为此可采用下面的策略来实现:(1)增加吸附态氧物种的碱性,如添加碱金属K等;(2)增加R-H中H原子的酸性(R为吸电子基团);(3)考虑空间位阻效应等因素,构造限制性Lewis酸碱对(Frustrated Lewis pairs (FLPs))[40],避免酸碱对(M-O)之间的强相互作用,以便更好地达到活化A-H键的目的.
猜你喜欢
发明与创新(2023年4期)2023-01-10
资源节约与环保(2022年8期)2022-09-20
椰城(2021年12期)2021-12-10
军民两用技术与产品(2021年10期)2021-03-16
少儿科学周刊·少年版(2021年22期)2021-01-17
水上消防(2020年1期)2020-07-24
疯狂英语·新读写(2018年3期)2018-11-29
现代营销(创富信息版)(2018年9期)2018-09-03
消费导刊(2017年20期)2018-01-03
消费导刊(2017年20期)2018-01-03
