
生物大分子高分辨率冷冻电镜三维重构技术
2018-07-03 07:10:48程凌鹏
实验技术与管理 2018年6期
程凌鹏
(清华大学 生命科学学院 蛋白质研究技术中心, 北京 100084)
冷冻电子显微三维重构技术(简称冷冻电镜)是20世纪80年代发展起来的一种透射电镜技术,能够解析生物大分子复合体的高分辨率三维结构。其特点是能够解析的生物样品的尺度较为宽泛,而且不需要样品结晶,保持了样品的近生理状态。冷冻电镜的这些特点与其他结构解析方法相比优势明显。X射线晶体学可以获得生物大分子原子分辨率的三维结构,但其局限在于需要先获得样品的结晶。而对于分子量较大、较复杂的生物大分子复合体通常难以得到结晶的样品,此外样品在结晶后的结构可能会发生变化。核磁共振虽然能解析溶液中的蛋白结构,但是只能解析分子量在50 kD以下的小分子结构。由于近年来随着电镜硬件和计算机图像处理技术的飞速发展,冷冻电镜三维结构解析分辨率从最初的纳米级推进到原子分辨率水平,一大批之前不能解析的生物大分子复合体的结构一下子变得可解,因此受到了生物学家的广泛关注,逐渐成为生物大分子结构解析的主流方法之一。2017年诺贝尔化学奖授予了开创冷冻电镜测定溶液中生物大分子复合体的高分辨率结构方法学的迪波什(Jacques Dubochet)、弗兰克(Joachim Frank)和亨德森(Richard Henderson)3位科学家。本文介绍了冷冻电镜高分辨率结构解析技术的起源、发展和未来趋势。
1 冷冻电镜技术的起源
1932年,德国物理学家鲁斯卡(Ernst Ruska)和克诺尔(Max Knoll)首次研制出了世界上第一台只有2个磁透镜的电子成像装置并将其命名为电子显微镜(简称电镜)。该装置最初的成像方法倍数只有十几倍。此后,鲁斯卡等人进一步改进了成像放大倍数和分辨率。他们的工作奠定了现代透射电镜的基础。因为电子的波长远小于可见光,所以电镜的分辨率远大于光学显微镜,可以达到0.1 nm左右。鲁斯卡也因此获得1986年的诺贝尔物理学奖[1]。在第一台电镜在实验室问世短短几年后,首台商业电镜产品就被生产出来并很快在材料科学领域获得了广泛的应用。
由于样品制备上的原因,电镜在生物领域的应用晚于材料科学。为了避免电子和空气分子发生碰撞散射,电镜必须在高真空下工作。而活性生物样品含有水分,真空会使其脱水从而破坏原有结构。此外,电子辐射也会破坏生物样品化学键导致结构破坏。因此,如何尽可能地保持生物样品的在电镜观察时的原有结构不发生变化是个问题。最早引入电镜制样的方法是负染。该方法用重金属包裹样品表面然后脱水干燥。这种方法虽然部分地解决了生物样品的电镜制样问题并被生物学家广泛地应用于生物样品的观察,但是其局限在于:(1)重金属本身有可能会破坏样品结构;(2)电镜观察到的只是样品的表面形貌;(3)染料不可能完全均匀地分布在样品表面。这些问题严重地限制了电镜图像分辨率。为了解决这些问题,格莱泽(Robert Glaeser)等人最早提出了利用冷冻的方法来降低样品辐射损伤[2-3],但是他们用传统方法获得的晶态冰会导致生物样品原有结构因为冰晶的产生而被破坏。迪波什和合作者在此基础上做了改进,他们将溶液中的样品迅速投入到液态乙烷中,样品溶液被快速冷冻形成非晶态冰,然后将样品放到液氮冷却的电镜冷台上观察。这样既能保持生物样品在冰中的原有结构,又避免了生物样品在真空中脱水,并且在一定程度上降低了辐射损伤[4-5]。迪波什等人的工作标志着冷冻电镜技术的诞生,这个方法至今仍被沿用。
2 冷冻电镜结构解析的基本原理
2.1 样品制备和电镜成像
冷冻电镜样品的具体制作流程如图1所示。首先,将高浓度和纯度的生物大分子溶液样品约4 μL加载到事先用亲水化处理过的铺有多孔碳膜的电镜载网上,然后用滤纸吸掉多余的溶液,使得只剩下一层几十到上百nm厚度的溶液薄膜留在碳膜上面。将该电镜载网迅速投入到用液氮冷却的液态乙烷中,这样溶液被迅速冷冻,形成非晶态的薄冰。从而避免了冰晶的形成对生物样品结构的破坏。不直接用液氮,而是乙烷,因为样品和液氮接触会迅速沸腾,产生的氮气隔在样品和液氮之间阻碍了热传导,从而形成晶态冰破坏样品结构。电镜图像从碳膜上的孔内的薄冰层中采集。为了防止过度剂量的电子辐射对样品的破坏,冷冻电镜采用低电子剂量的成像方式。因为样品纯度很高,所有样品颗粒的结构是均一的(具有相同的三维结构),而且冰中的样品颗粒是随机取向,每个样品颗粒的电镜图像可以看作是单个颗粒从不同方向上的投影。颗粒的三维结构可以通过计算机重构的方法合并这些投影图得到。因此,在冷冻电镜制样前用生化方法获得结构均一的样品非常重要。
由于冷冻电镜样品非常薄,而且主要由碳、氢和氧等轻原子构成,因此能满足弱相位体近似(weak-phase object approximation),也就是说,可以认为入射电子通过样品只发生一次弹性散射,振幅不变,相位变化很小。由于构成生物样品的原子的原子量和水,接近以及成像采用较低的电子剂量,所以在正焦条件下电镜图像的衬度很小。因此样品的图像衬度主要通过成像时一定程度的离焦(通常是欠焦)来实现。由于图像的欠焦和物镜的球差效应,冷冻电镜图像没有完全真实地反映样品颗粒的投影,而是傅里叶空间被衬度传递函数(CTF)所调制的投影图(见图2)。CTF曲线可以写为空间频率S的函数[6]:
CTF(S)=-sin(γ(S)+Φ)
(1)
其中γ(S)=π((-Csλ3S4)/2+ΔzλS2),S是空间频率,Q是振幅衬度在整个图像衬度中所占的比例,由样品厚度和电子能量所决定,在冷冻电镜成像中约为0.1弧度;Cs是物镜球差系数;λ是电子波长,由电镜加速电压决定;Δz是成像时的离焦值,由用户给定。
图2(a)是一张乳糖苷酶的电镜图像[7],图2(b)是冷冻电镜图像的傅里叶变换图,图2(c)是CTF函数图。从图中可以看到,图像的低频部分只是有衬度反转,而高频信息则被完全扭曲。因此要从电镜图像中获得高分辨率的生物样品的投影图像,必须先对图像进行CTF校正。
2.2 电镜图像的计算机三维重构
生物样品电镜图像三维重构在冷冻电镜技术出现之前已经有人研究。1968年,克鲁格(Aaron Klug)和德罗西耶(David DeRosier)用三维重构的方法合并多张噬菌体螺旋对称的尾部负染电镜图像获得了它的三维结构[8]。重构算法的基础是中央截面定理:一个三维物体的二维投影的傅里叶变换是该物体的三维傅里叶变换中的垂直于投影方向的一个中央截面。如果可以获得该物体在全空间中不同方向的投影,然后对每张投影图进行傅里叶变换,并按投影方向填充到三维傅里叶空间对应的中央截面,并且如果投影的空间取向足够多且均匀分布,那么其数据点就可以布满该三维物体的整个三维傅里叶空间。对于三维傅里叶空间的空隙的部分可以通过插值计算得到。对该三维物体的傅里叶空间进行三维反傅里叶变换就可得到物体实空间的三维结构。此后,1971年克劳瑟(Richard Crowther)用类似的方法解析了首个二十面体对称病毒衣壳的结构[9]。1975年亨德森等人首次用电镜解析了细菌紫膜蛋白的二维晶体结构,并在1990年首次将推进到近原子分辨率水平[10-11],证明了电镜结构解析分辨率的潜力。因为在生物大分子复合体电镜图像三维重构领域的开创性贡献,克鲁格获得了1982年诺贝尔化学奖。
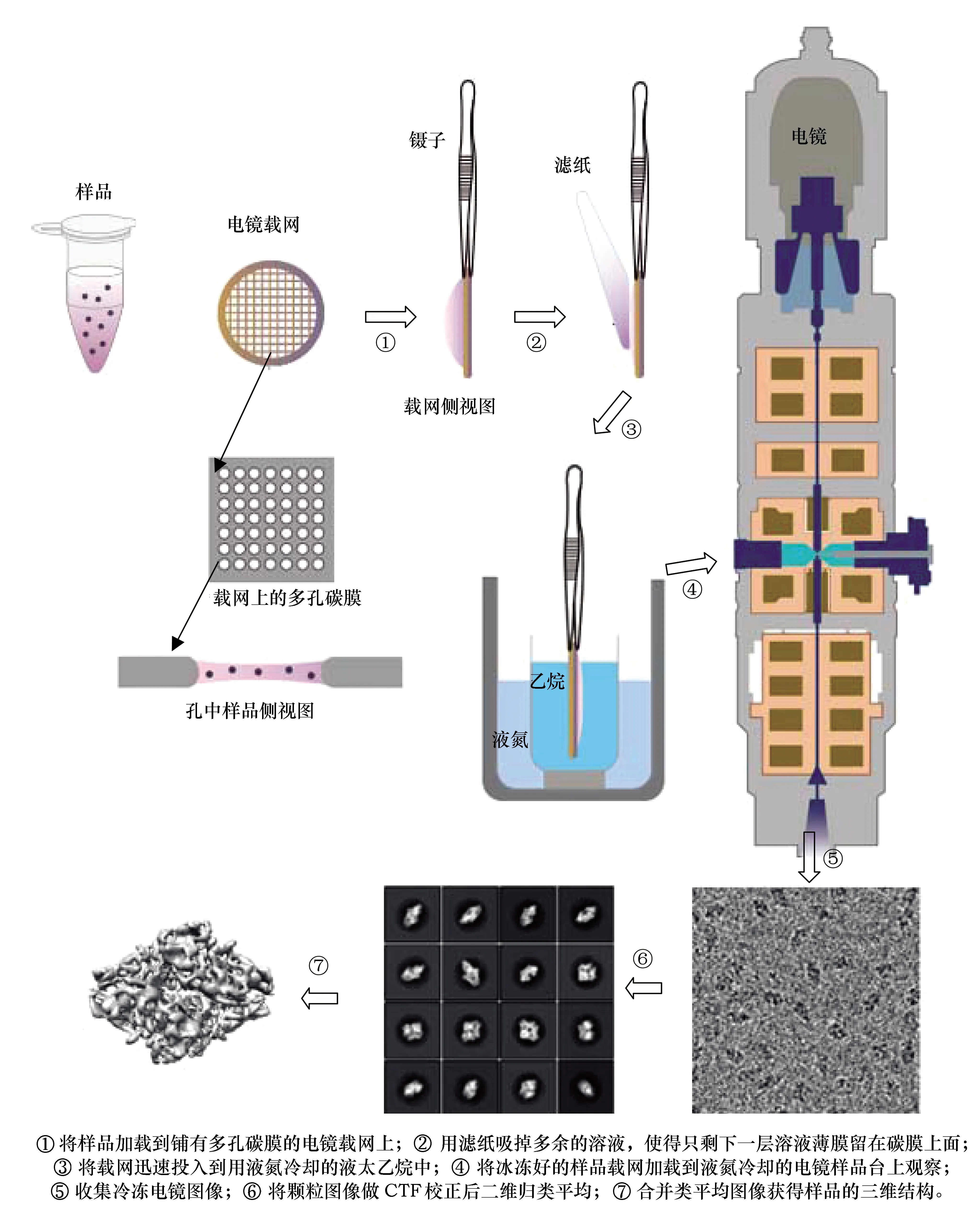
图1 冷冻电镜样品制作流程图
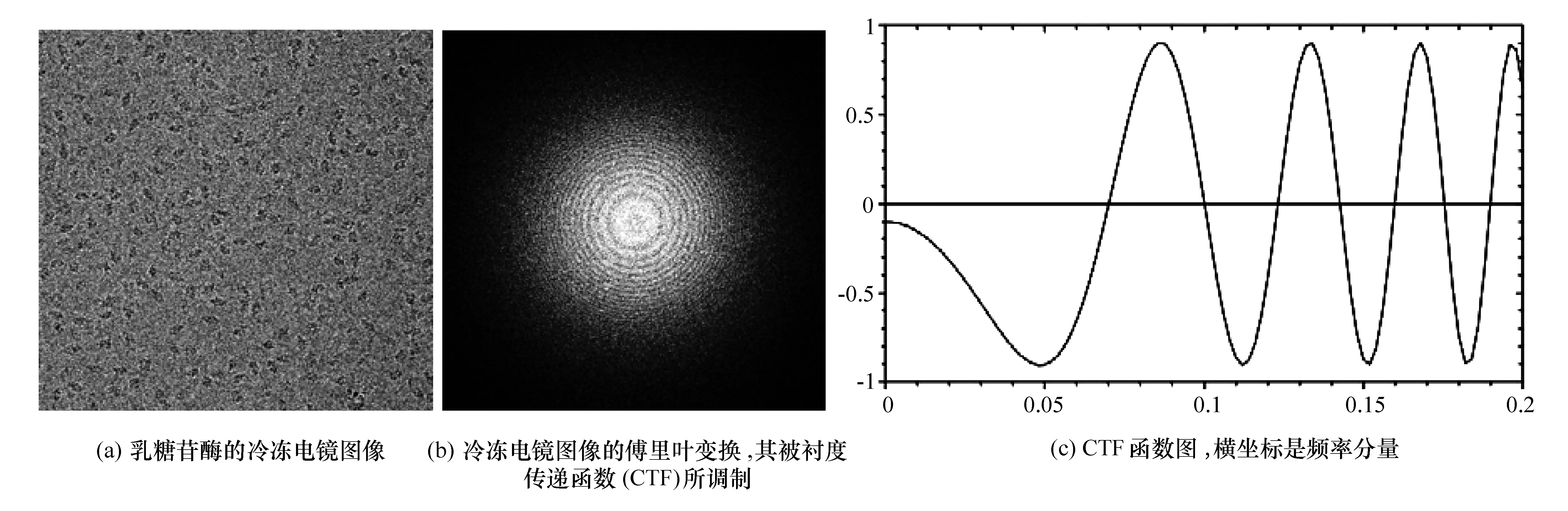
图2 冷冻电镜图像及CTF函数图
以上电镜三维重构的共同特点是都利用了样品的高对称性或样品形成有序排列的二维晶体。然而,多数生物大分子复合体没有对称性且难以形成二维晶体。1974年,弗兰克针对这个问题提出通过对框取出的样品颗粒图像按照其相关性做分类,然后将相同类的图像做二维平均以提高信噪比[12]。每一个类图像平均代表了样品颗粒在某个方向的投影图,合并这些投影图就可以获得三维结构[13]。冷冻电镜图像三维重构的流程如下:
(1) 通过取向参数拟合的方法测定每一张图像的欠焦值和像散并对图像进行CTF校正。
(2) 获得初始结构模板。颗粒结构的低分辨率初始结构可以采用与样品同源的已知结构;如果没有类似结构,就需要用收集样品的倾转图像来获得初始结构模板[14]。
(3) 把样品颗粒图像从电镜图像中框取出来并按照图像的相似度进行归类,对同一类的图像进行平均,得到类平均图像。
(4) 用投影匹配方法获得每个类平均图像的空间取向和中心,投影匹配法需要一个初始结构作为参考模板,程序首先对这个初始结构做全空间取向投影作为模板,然后将每张类平均图像和这些投影模板用交互相关的方法做相似度比较,每张类平均图像中的颗粒的空间取向被测定为与其相似度最大的投影模板的投影取向。
(5) 合并所有的被测定了空间取向和中心位置的类平均图像进行三维重构。重构算法除了上面所说的傅里叶空间插值法外,还有反投影重构法[15]。
(6) 在得到了新的三维结构后,将该三维结构作为新的模板,重新匹配所有的颗粒。
(7) 循环迭代第(5)—(6)步,并不断提高重构分辨率。最终的有效结构分辨率通过计算两组独立处理的颗粒图像的重构结果的傅里叶变换的径向相关性来获得[16]。
3 冷冻电镜技术发展
随着电镜硬件和软件技术的不断改进和提高,用冷冻电镜技术解析的生物复合体结构的分辨率也在不断提高。最初的三维重构分辨率只有2~4 nm[17-18]。在众多非二维晶体的生物样品中,由于二十面体病毒具有高度对称性,总是最早获得结构分辨率突破的大分子复合体。1997年,乙肝病毒的亚纳米分辨率冷冻电镜结构首次被测定。在0.7~0.9 nm的分辨率下,首次观察到乙肝病毒衣壳蛋白中的α螺旋呈现棍状结构[19-20]。2008年,首个用基于冷冻电镜方法获得的病毒衣壳蛋白的骨架模型被构建[21]。此后低对称性和无对称性的复合体结构也获得了类似分辨率的结构[22-23]。近年来用冷冻电镜结构达到可以直接构建原子模型的分辨率水平[24-25]。最具有代表性的工作是2013年程亦凡研究组和朱利叶斯研究组合作,首次使用基于直接电子探测技术的相机收集冷冻电镜图像重构获得了大小仅为300 kD的一种辣椒素受体蛋白的0.34 nm分辨率的结构[26],表明冷冻电镜技术解析较小的蛋白复合体结构也能获得近原子分辨率水平。目前冷冻电镜技术所获得的结构分辨率已经突破0.2 nm,达到了X射线晶体学结构解析的分辨率水平[27]。
3.1 电镜和相关设备的发展促进了电镜图像质量和采集效率的提高
冷冻电镜结构解析分辨率的提高与技术的发展总是分不开的。电镜高分辨率技术的一个较早的突破是场发射电子枪(FEG)的出现。场发射电子枪和热电子枪相比能获得相干性更好的电子光源,因此能获得的图像的衬度和分辨率更高。平行入射电子光源的应用进一步提高了电子穿过样品后的相干性。磁透镜的恒定功率系统增加了透镜电流的稳定性。近年来开发的冷冻电镜图像自动收集软件的普及使得电镜可以在无人监督下24 h自动收集图像,大大地减轻了使用者的劳动量,提高了结构解析的效率。电镜样品自动加载系统(autoloader system)的出现很大程度上减少了样品的冰晶污染并增加了样品台的稳定性。冷冻样品自动制样机的推广使得电镜载网上冰的厚度可控性增加,大大提高了冷冻样品制备的成功率和可重复性。
3.2 冷冻电镜图像质量的提高
因为冷冻电镜成像的电子剂量低,且生物复合体的主要元素碳、氢和氧等原子量和构成水的原子量接近,所以冷冻电镜技术的最大的瓶颈是图像的衬度和信噪比低。电子辐照到样品上导致样品的抖动漂移进一步削弱了冷冻电镜图像的高分辨率信息。较低的电镜图像衬度和信噪比导致后期精确测定颗粒取向上的困难。直接电子探测技术(direct-electron detector device,DDD)的相机的使用极大地改善了冷冻电镜图像的质量。电镜图像采集器件的效率由电子转换效率(detective quantum efficiency,DQE)也就是输出信号和信噪比的平方和输入信号的信噪比的平方的比值来衡量[28]。在DDD相机出现以前,冷冻电镜图像通常用电荷耦合器件(charge coupled device,CCD)相机或者摄影底片来收集。CCD相机收集图像比底片要方便,而且在低频区域的DQE较高。但因为CCD不能够直接探测电子图像信号,而是要通过闪烁体将电子信号转换为光信号来获得电镜图像,在转换的过程中不可避免地引入了噪声,所以其高频信号的DQE较差。底片收集图像的高频区域的DQE虽然比CCD相机高[29],但是不能即时可见,而是需要后期显影和扫描才能获得数字图像,使用起来很不方便。DDD相机因为可以直接探测电子信号,所以在各个频率段的DQE都远高于底片和CCD相机,极大地提高了电镜图像的衬度和信噪比。近年来DDD相机已成为冷冻电镜图像采集的标配。
除了DQE值高外,DDD还有一个特点是能够以视频的形式记录,把一张图像分为每秒钟数十到数百帧的动画图像。DDD成像的这一特性使得在电镜成像时的样品漂移可以通过对这些帧图像进行配准后平均来校正[30-31]。这样就能减少或者避免因为样品漂移导致的图像模糊。DDD技术不仅提高了冷冻电镜成像质量使得用同样的数据量可以获得更高分辨率的三维结构,而且是对解析1 000 kD以下的较小蛋白复合体结构突破到原子分辨率水平的最大的技术推动。
3.3 生物样品三维分类和分辨率的提高
冷冻电镜三维重构的一个特点是通过合并大量样品颗粒图像的平均图实获得样品颗粒的三维结构,因而样品中含有少量其他不同结构的杂质颗粒不影响最后的三维重构结果。如果样品中所含不同结构的颗粒较多,尤其是同种样品的多个不同构象之间具有较小结构差异,那么这些因素会导致平均图像的模糊,最终影响重构分辨率的提高。生物样品中这类情况很常见,尤其是同种样品可能有不同的构象,每个构象都代表了样品的一个和其生物功能相关的状态。因此将不同构象的颗粒区分出来对研究其生物机制有重要意义。最大似然(maximum likelihood)方法应用在颗粒分类上能够识别颗粒图像究竟是某个颗粒的不同方向上的投影还是不同结构颗粒的投影[32]。通过这种方法,溶液中不同的颗粒可以按其三维结构的不同分为不同的类。这种分类也促进了重构分辨率的提高。
冷冻电镜及其相关设备的一系列技术的改进和图像三维重构软件发展是相辅相成的。电镜设备技术改进提高了图像的衬度和信噪比,反过来更好质量的图像也促进了三维重构图像处理软件的研发,这些因素共同推动了冷冻电镜生物大分子复合体结构解析分辨率的革命性突破[33-34]。
4 冷冻电镜技术的未来发展趋势
从1997年报道的首个解析到亚纳米分辨率的乙肝病毒的结构[19-20]到2016年报道的0.18 nm分辨率的谷氨酸脱氢酶(约300 kD)结构[27],冷冻电镜结构生物学在这20年来经历了飞速的发展。能够用冷冻电镜获得近原子分辨率水平的结构的样品范围从最初的大到病毒这样的超大分子复合物到现在的不到100 kD的小分子复合物[27],其解析小分子复合物的尺度范围已经部分覆盖到了原本只有X射线晶体学才能达到的范围。目前冷冻电镜结构解析技术已经非常成熟,只要有合适的大小、浓度和结构均一性的样品,单纯从结构解析技术上来说,要获得样品的高分辨率结构已经没有挑战性可言。但是在适用样品的尺度普适性这一问题上依然有很多技术问题有待解决。例如,颗粒尺度太小会导致电镜图像的衬度低,在数据处理时难以准确地匹配图像,目前仍然只有少数100 kD左右的小分子复合体用冷冻电镜方法能够获得近原子分辨率的结构。此外,由于电镜的场深有限,要解析较大的颗粒的样品的原子分辨率结构仍然是一个挑战。要让冷冻电镜高分辨率结构技术能够适用更多的生物样品,使结构解析变得更为普适,永远是摆在冷冻电镜方法学研究者面前的首要课题。
4.1 相位板技术为解析小分子结构提供了可能
从成像原理上,冷冻电镜图像衬度低的原因是CTF函数的正弦函数取向调制导致图像低频部分被衰减,如前面公式(1)和图2C所示。因此目前用冷冻电镜解析小分子,尤其是100 kD以下的小分子的高分辨率结构依然是一个挑战。如果可以通过某种手段使得公式(1)中的相移Φ变为π/2,那么该正弦函数则变为余弦函数CTF(S)=-cos(γ(S)),图像的低频部分就不存在衰减,衬度就会显著增强。近年来相位板(phase plate)技术的出现解决了这个问题[35]。相位板是加在电镜物镜后焦面上的一层特殊的薄碳膜,能够实现这个相移。由于图像衬度的增强,可以用正焦或者很小的欠焦,甚至小的过焦成像[36],这样也避免了因为欠焦过大导致的图像信息的高频衰减。尽管相位板技术在成像稳定性方面有待进一步提升,但是该技术为目前冷冻电镜高分辨率三维重构技术突破小分子尺度极限提供了可能。
4.2 埃瓦尔德校正技术是解析超大分子复合体的原子分辨率结构的方向
在冷冻电镜结构高分辨率解析的另一个极端,如果生物样品的尺度很大(50 nm以上),要获得其超过0.3 nm分辨率的结构也很困难。在冷冻电镜图像处理时,通常忽略样品的厚度。如果样品的厚度小于电镜的场深,这个近似是合理的。但是如果样品的厚度过大,必然导致样品沿着入射电子方向上不同层面的欠焦值不同,从而导致图像高分辨率信息由于欠焦值差异而混叠。考虑到这一点,每张样品颗粒的冷冻电镜图像的傅里叶变换就不再是对应于穿过原点的中央截面,而是穿过原点的一个球形曲面,这样的电镜图像校正称为埃瓦尔德(Ewald)校正[37]。当分辨率要求不高且颗粒尺度很小的时候,该曲面和截面可认为是近似相同的。这就解释了尽管二十面体病毒样品是在所有生物样品中首个获得0.3~0.4 nm分辨率结构的样品,但是分辨率很难继续提高了。由于冷冻电镜图像信噪比较低且埃瓦尔德校正算法复杂,目前该校正并没有被广泛地应用。要推进较大颗粒的重构分辨率,电镜图像的埃瓦尔德校正有待进一步研究。
4.3 某些样品在溶液中的优势取向问题有待解决
冷冻电镜样品中的优势取向也会影响重构分辨率。如之前提到的,冷冻电镜三维重构的一个前提就是需要得到颗粒图像的空间取向足够多而且均匀分布。但是有一些样品在溶液中的空间取向并不均匀,而是在某些取向的颗粒极多,其他取向很少。这种情况严重影响最后的重构分辨率。尽管目前解决这类问题有一些可选方案[38],但是这些方案并非对所有样品都有效,或者会对成像质量产生负面影响。因此,如何克服溶液中生物样品颗粒的优势取向是一个值得研究的课题。
4.4 建立若干个冷冻电镜设备和技术资源集中的共享科研设施是大势所趋
目前建立一个300 kV的主流冷冻电镜平台需要数千万元人民币的设备购置开支,冷冻电镜设备的昂贵的价格和维护成本使得大多数科研单位无法建立这样的结构解析平台。而且,并不是所有的生物学家都是冷冻电镜专家,他们只关注和结构相关的生物样品的分子机制。冷冻电镜维护和操作的复杂性也提高了生物学家的结构解析技术门槛。因此,建设若干个科研共享的冷冻电镜设施显得尤为重要。冷冻电镜结构解析的最终发展的趋势是和X射线晶体学结构解析共享科研平台一样,生物学家只要将合适的生物样品交给结构解析技术平台,就能得到样品的高分辨率三维结构。冷冻电镜结构解析技术的发展必定会让生物学家更专注于生物学,而不是结构解析技术本身。
[1] Williams D B. Carter C B, Transmission electron microscopy: a textbook for materials science[M]. Springer-Verlag:1996.
[2] Taylor K A, Glaeser R M. Electron-Diffraction of Frozen, Hydrated Protein Crystals[J]. Science, 1974 (186):1036-1037.
[3] Taylor K A, Glaeser R M. Electron microscopy of frozen hydrated biological specimens[J]. J Ultrastruct Res, 1976 (55):448-456.
[4] Dubochet J, Mcdowall A W. Vitrification of Pure Water for Electron-Microscopy[J]. Journal of Microscopy-Oxford, 1981 (124):3-4.
[5] Adrian M, Dubochet J, Lepault J, et al. Cryo-electron microscopy of viruses[J]. Nature, 1984 (308):32-36.
[6] Erickson H P, Klug A. Fourier Transform of an Electron Micrograph-Effects of Defocussing and Aberrations, and Implications for Use of Underfocus Contrast Enhancement[J]. Berichte Der Bunsen-Gesellschaft Fur Physikalische Chemie, 1970(74):1129-1132.
[7] Bartesaghi A, Merk A, Banerjee S, et al. A resolution cryo-EM structure of beta-galactosidase in complex with a cell-permeant inhibitor[J]. Science, 2015 (348):1147-1151.
[8] Derosier D J, Klug A. Reconstruction of 3 Dimensional Structures from Electron Micrographs[J]. Nature, 1968 (217):130-134.
[9] Crowther R A. Procedures for three-dimensional reconstruction of spherical viruses by Fourier synthesis from electron micrographs[J]. Philos Trans R Soc Lond B Biol Sci, 1971 (261):221-230.
[10] Henderson R, Unwin P N T. 3-Dimensional Model of Purple Membrane Obtained by Electron-Microscopy[J]. Nature, 1975 (257):28-32.
[11] Henderson R, Baldwin J M, Ceska T A, et al. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy[J]. J Mol Biol, 1990 (213):899-929.
[12] Frank J. Averaging of low exposure electron micrographs of non-periodic objects[J]. Ultramicroscopy, 1975 (1):159-162.
[13] Frank J, Shimkin B, Dowse H, Spider-a Modular Software System for Electron Image-Processing[J]. Ultramicroscopy, 1981 (6):343-357.
[14] Radermacher M, Wagenknecht T, Verschoor A, et al. Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli[J]. J Microsc, 1987 (146):113-136.
[15] Frank J. Three-dimensional electron microscopy of macromolecular assemblies[M].Oxford University Press:2006.
[16] Chen S, McMullan G, Faruqi A R, et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy[J]. Ultramicroscopy, 2013 (135):24-35.
[17] Frank J, Zhu J, Penczek P, et al. A model of protein synthesis based on cryo-electron microscopy of the E. coli ribosome[J]. Nature, 1995 (376):441-444.
[18] Frank J, Penczek P, Grassucci R, et al. Three-dimensional reconstruction of the 70S Escherichia coli ribosome in ice: the distribution of ribosomal RNA[J]. Cell Biol, 1991 (115):597-605.
[19] Conway J F, Cheng N, Zlotnick A, et al. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy[J]. Nature, 1997 (386):91-94.
[20] Bottcher B, Wynne S A, Crowther R A. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy[J]. Nature, 1997 (386):88-91.
[21] Jiang W, Baker M L, Jakana J, et al. Backbone structure of the infectious epsilon 15 virus capsid revealed by electron cryomicroscopy[J]. Nature, 2008 (451):1130-1134.
[22] Ludtke S J, Baker M L, Chen D H, et al. De novo backbone trace of GroEL from single particle electron cryomicroscopy[J]. Structure, 2008 (16):441-448.
[23] Hashem Y, des Georges A, Fu J, et al. High-resolution cryo-electron microscopy structure of the Trypanosoma brucei ribosome[J]. Nature, 2013 (494):385-389.
[24] Zhang X, Jin L, Fang Q, et al. Angstrom Cryo-EM Structure of a Nonenveloped Virus Reveals a Priming Mechanism for Cell Entry[J]. Cell, 2010 (141):472-482.
[25] Cheng L, Sun J, Zhang K, et al. Atomic model of a cypovirus built from cryo-EM structure provides insight into the mechanism of mRNA capping[J]. Proc Natl Acad Sci USA, 2011 (108):1373-1378.
[26] Liao M, Cao E, Julius D, et al. Structure of the TRPV1 ion channel determined by electron cryo-microscopy[J]. Nature, 2013 (504):107-112.
[27] Merk A, Bartesaghi A, Banerjee S, et al., Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery[J]. Cell, 2016 (165):1698-1707.
[28] McMullan G, Faruqi A R, Clare D, et al. Comparison of optimal performance at 300keV of three direct electron detectors for use in low dose electron microscopy[J]. Ultramicroscopy, 2014 (147):156-163.
[29] Booth C R, Jakana J, Chiu W. Assessing the capabilities of a 4kx4k CCD camera for electron cryo-microscopy at 300kV[J]. J Struct Biol, 2006 (156):556-563.
[30] Brilot A F, Chen J Z, Cheng A, et al. Beam-induced motion of vitrified specimen on holey carbon film[J]. J Struct Biol, 2012 (177):630-637.
[31] Li X, Mooney P, Zheng S, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM[J]. Nat Methods, 2013 (10):584-590.
[32] Scheres S H, Gao H, Valle M, et al. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization[J]. Nat Methods, 2007 (4):27-29.
[33] Kuhlbrandt W, Biochemistry. The resolution revolution[J]. Science, 2014 (343):1443-1446.
[34] Bai X C, McMullan G, Scheres S H. How cryo-EM is revolutionizing structural biology[J]. Trends Biochem Sci, 2015 (40):49-57.
[35] Danev R, Buijsse B, Khoshouei M, et al. Volta potential phase plate for in-focus phase contrast transmission electron microscopy[J]. Proceedings of the National Academy of Sciences of the United States of America, 2014 (111):15635-15640.
[36] Fan X, Zhao L Y, Liu C, et al. Near-Atomic Resolution Structure Determination in Over-Focus with Volta Phase Plate by Cs-Corrected Cryo-EM[J]. Structure, 2017 (25):1623-1628.
[37] Wolf M, DeRosier D J, Grigorieff N. Ewald sphere correction for single-particle electron microscopy[J]. Ultramicroscopy, 2006 (106):376-382.
[38] Cheng Y, Grigorieff N, Penczek P A, et al. A primer to single-particle cryo-electron microscopy[J]. Cell, 2015 (161):438-449.
猜你喜欢
摄影世界(2022年1期)2022-01-21 10:50:14
数学物理学报(2019年3期)2019-07-23 01:15:40
知识经济·中国直销(2018年12期)2018-12-29 12:22:14
家庭影院技术(2018年9期)2018-11-02 05:31:32
商周刊(2017年6期)2017-08-22 03:42:36
自动化学报(2017年5期)2017-05-14 06:20:52
成都信息工程大学学报(2017年6期)2017-03-16 03:04:32
河北林业科技(2016年5期)2016-11-08 03:12:48
山东大学法律评论(2016年0期)2016-08-16 03:24:12
华东理工大学学报(自然科学版)(2015年3期)2015-11-07 09:17:35
