
液相色谱法及质谱法测定柴油芳烃适用性研究
2018-06-05 11:17:13张祎玮祝馨怡刘泽龙徐广通
石油炼制与化工 2018年6期
张祎玮,祝馨怡,刘泽龙,徐广通
(中国石化石油化工科学研究院,中国石化分子炼油重点实验室,北京 100083)
随着对移动污染源排放限值的日益严格,车用燃料的质量标准也不断升级。在2017年全国实施国Ⅴ排放标准的基础上,2018年东部部分省份又将实施更加严格的国Ⅵ排放标准。柴油作为重要的车用燃料,其中的多环芳烃类化合物不仅直接影响柴油的十六烷值及燃烧性能,也会使尾气排放中的固体颗粒物、氮氧化合物等污染物含量升高[1-2],柴油中的多环芳烃含量一直是柴油质量升级的重要指标。柴油中芳烃及多环芳烃的准确测量对炼油工艺及催化剂的研究以及柴油产品的质量控制与质量评定具有重要意义。早在20世纪70年代起,色谱技术就开始应用于石油产品组分分析[3-6],高效液相色谱和质谱是测定柴油中芳烃含量的重要手段。SH/T 0806—2008标准[7]适用范围为150~400 ℃馏分油,采用液相色谱,利用极性柱分离芳烃和非芳烃,并根据环结构分离为单环、双环和三环及以上芳烃,使用示差折光检测器检测,外标法定量,可测定单环、双环、三环及三环以上芳烃含量,该标准方法等效采用了ASTM D6591-06标准[8]。基于气相色谱质谱联用技术的SH/T 0606—2005标准[9]适用范围为204~365 ℃柴油馏分,采用固相萃取法将试样分离成饱和烃和芳烃后分别进行质谱测定。根据特征质量碎片加和确定各类烃的浓度,可测定11类烃组成,该标准方法等效采用了ASTM D2425-04标准[10]。本研究基于SH/T 0806—2008和SH/T 0606—2005方法,对比采用高效液相色谱示差折光检测器(HPLC)和气相色谱-质谱联用(GC-MS)测定柴油中芳烃含量的分析结果、加标回收率及掺兑测定的准确性,讨论两种方法分析结果的差异性和适用性,并在此基础上对8个国Ⅵ车用柴油样品的芳烃含量进行测定。
1 实 验
1.1 仪器与原料
美国Agilent公司生产的HPLC1260型高效液相色谱仪,配备示差折光检测器,色谱柱采用美国Waters公司生产的Spherisorb氰基键合正相色谱柱,固定性填充颗粒直径5 μm,内径4.6 mm,柱长 250 mm,柱温箱内配有六通阀;美国Agilent公司生产的6890GC-5975MS气相色谱质谱联用仪。
环己烷、邻二甲苯,分析纯,北京精细化学品有限公司生产;四氢萘、2-甲基联苯,分析纯,TCI公司生产;1-甲基萘、菲、二苯并噻吩、9-甲基蒽、芴,均为分析纯,百灵威公司生产;正庚烷,色谱纯,伊诺凯公司生产。
试验用原料油包括:直馏柴油Ⅰ、Ⅱ,记为柴油1和柴油2;催化裂化柴油Ⅰ、Ⅱ,记为柴油3和柴油4;0号国Ⅲ、国Ⅴ和国Ⅵ成品车用柴油,记为柴油5、柴油6和柴油7;加氢精制柴油Ⅰ,Ⅱ,Ⅲ,记为柴油8、柴油9、柴油10;掺兑实验选用的加氢裂化柴油为几乎不含有多环芳烃的柴油样品,记为柴油11;选用的直馏柴油记为柴油12;国Ⅵ车用柴油样品芳烃含量对比分析所选用的8个样品分别编号为国Ⅵ-1~国Ⅵ-8。
1.2 高效液相色谱分析
HPLC法参考SH/T 0806—2008方法,柱温箱中安装反冲洗阀,使得示差折光检测器可独立于色谱柱内液体的流向,通过HPLC分析可得到单环芳烃、双环芳烃、三环及三环以上芳烃含量,进样量10 μL,流动相流速0.8 mL/min,柱温30 ℃,示差折光检测器温度30 ℃。
1.3 气相色谱-质谱联用
GC-MS参考SH/T 0606—2005方法,采用100%二甲基聚硅氧烷非极性色谱柱(30 m×250 μm×0.25 μm),进样量0.5 μL,分流比20∶1,气化温度300 ℃,GC柱箱温度80 ℃,保持3 min,再以40 ℃/min速率升温至300 ℃,保持5 min,载气流速1.0 mL/min,GC-MS接口温度300 ℃,EI源电离方式,电子能量70 eV,离子源温度250 ℃,检测质荷比(m/z)范围为50~500。
2 结果与讨论
2.1 液相色谱法与质谱法分析结果对比
选取柴油1~柴油9,根据SH/T 0806—2008和SH/T 0606—2005方法,分别采用HPLC及GC-MS法分析其中单环芳烃、双环芳烃、三环及三环以上芳烃含量,结果见表1。从表1可以看出:HPLC法测定的总芳烃和单环芳烃含量普遍比GC-MS法测定的总芳烃和单环芳烃含量高;对于大部分柴油如直馏柴油、成品柴油,HPLC法测定的双环芳烃含量与GC-MS结果相比普遍偏低;三环及三环以上芳烃含量均与GC-MS测定结果相近;多环芳烃(双环芳烃、三环及三环以上芳烃总和)含量普遍比用GC-MS测定结果偏低,但对于催化裂化柴油以及一些进行过加氢处理的柴油,其双环芳烃总含量明显较高,对于这类油,HPLC法测定的多环芳烃含量相较于质谱测定结果则会出现不同程度的偏高。

表1 采用HPLC和GC-MS法测定不同类型柴油中芳烃含量的结果对比 w,%
注:HPLC简写为LC,GC-MS简写为MS。表6同。
质谱法对于总芳烃含量的测定是基于层析色谱法对柴油的饱和烃和芳烃进行分离,可确定总芳烃的含量,对分离得到的饱和烃和芳烃进行GC-MS分析,可进一步修正层析色谱法分离得到的饱和烃和芳烃之间的交叉造成的误差,从而保障质谱法测得总芳烃含量的准确性。HPLC法测定的总芳烃含量与GC-MS法相比普遍偏高,这主要是由于液相色谱测定柴油中单环芳烃明显偏高造成的,特别是对于单环芳烃含量较高的样品,定量误差甚至会高达15%。
2.2 方法准确性及回收率
为考察两种方法自身分析结果的准确性,将加氢裂化柴油(柴油11)与直馏柴油(柴油12)进行掺兑,研究两种方法的掺兑准确性。将两种柴油分别按质量比1∶1和2∶1进行掺兑,使用HPLC和GC-MS法测定芳烃含量,结果见表2。由表2可以看出:①柴油11中几乎不含有任何多环芳烃,将其与柴油12进行质量比1∶1掺兑后,HPLC测得单环芳烃、双环芳烃、三环及三环以上芳烃质量分数分别为7.7%,4.2%,0.3%,与理论计算值7.8%,4.3%,0.3%的偏差在0~2%之间;②GC-MS法测得单环芳烃、双环芳烃、三环及三环以上芳烃质量分数分别为8.6%,4.6%,0.2%,与理论计算值9.0%,4.6%,0.2%的偏差在0~5%之间;③对于以质量比2∶1掺兑情况,两种方法实验测得单环芳烃、双环芳烃、三环及三环以上芳烃含量的实际值与理论值接近,偏差均小于6%。说明两种方法对于柴油掺兑后芳烃含量测定的准确性均较好。

表2 采用HPLC和GC-MS法测定掺兑柴油中芳烃含量的结果对比 w,%
注:括号内为理论计算值。
2.3 模型化合物对定量结果准确性的影响
实际柴油样品中,单环芳烃主要为烷基苯类和四氢萘类形式,也有CnH2n-10(如茚及八氢菲类等);双环芳烃主要为萘类、CnH2n-14(如联苯类、苊类)及CnH2n-16(如芴类)结构;三环芳烃主要为CnH2n-18(如菲类和蒽类)结构。液相色谱法分离得到的谱图中,单环芳烃、双环芳烃、三环及三环以上芳烃3个大峰实际上是由多种结构相似而折射率不同的组分构成的混合峰。由于示差折光检测器对不同物质的响应具有不均一性,而不同柴油中芳烃组成又很复杂,当进行族组成定量时,采用一种标准化合物代表一类化合物的响应因子定量误差较大,因此需选择更为合适的标准化合物乃至模型油以提高柴油中各类芳烃定量的准确性。
2.3.1单环芳烃模型化合物研究SH/T 0806—2008方法采用邻二甲苯作为单环芳烃标准化合物。柴油中的单环芳烃除烷基苯类化合物外,还有大量的四氢萘类和CnH2n-10化合物,特别是以催化裂化柴油为原料通过加氢生产的柴油。由于烷基苯类和四氢萘类化合物的折射率存在差别,随着柴油中单环芳烃中四氢萘类化合物含量的逐渐增加,使用邻二甲苯作为标准化合物时,单环芳烃测定结果的误差会逐渐增大[11]。
采用不同单环芳烃模型化合物时HPLC与GC-MS法分析结果偏差见表3。为进一步探究单环芳烃标准物选择对于测定结果误差的影响,选择四氢萘类明显高于烷基苯含量的加氢柴油进行实验,表3中给出了它们的实际值,这些样品的单环芳烃含量较高,且单环芳烃中四氢萘或茚满类化合物含量也较高。因此,采用四氢萘作为标准物重新绘制工作曲线进行单环芳烃含量测定。从表3可以看出,采用四氢萘作为标准化合物时测定的单环芳烃含量误差明显减小,同时对几种成品油单环芳烃响应因子进行考察,结果发现,同样随着四氢萘类化合物在柴油中含量的增加,使用邻二甲苯作为标准化合物进行测定的误差会逐渐增大,而此时若换用四氢萘作为标准化合物,测定误差可以显著降低。
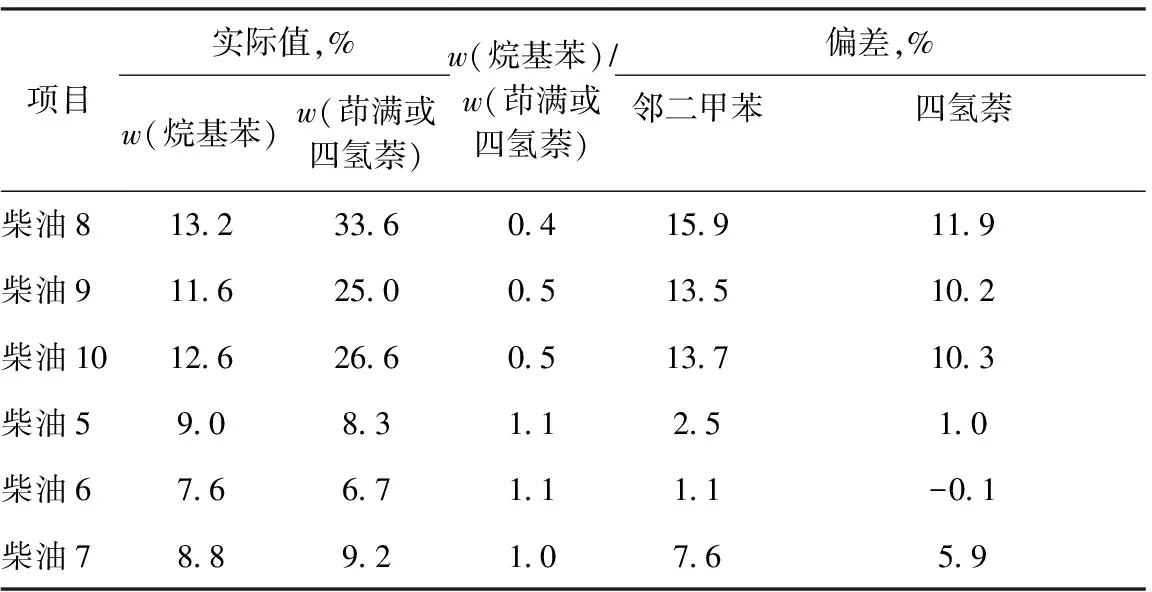
表3 采用不同单环芳烃模型化合物时HPLC与GC-MS法分析结果偏差
进一步分析邻二甲苯和四氢萘的折射率可知,四氢萘的折射率大于邻二甲苯的折射率,在使用示差折光检测器检测所得的工作曲线斜率也会大于邻二甲苯。图1为分别以邻二甲苯和四氢萘为模型化合的单环芳烃工作曲线对比,两条工作曲线的线性系数均为0.999,因此当单环芳烃峰面积一定时,用四氢萘作为模型化合物测量所得的单环芳烃含量会偏小。
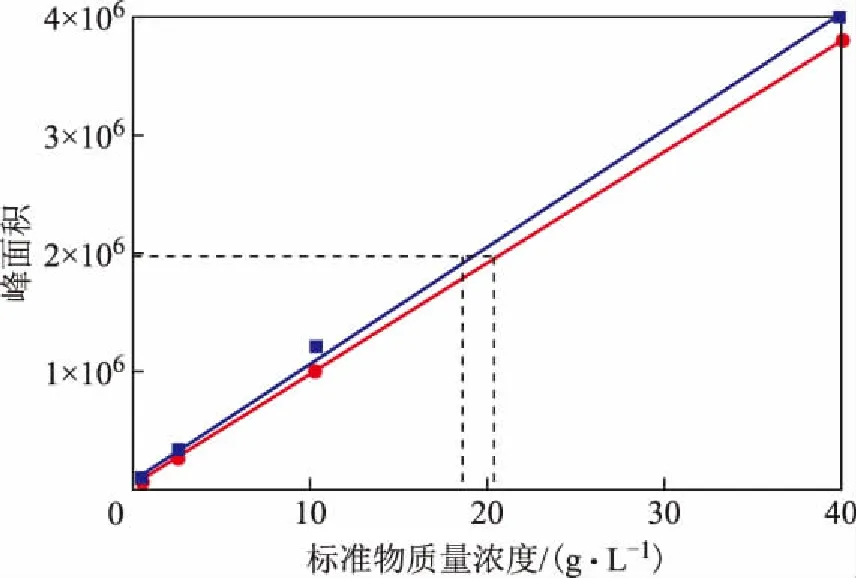
图1 不同单环芳烃模型化合物的工作曲线对比●—邻二甲苯; ■—四氢萘
2.3.2双环芳烃定量加标准确性考察SH/T 0806—2008方法采用1-甲基萘作为双环芳烃标准化合物。柴油中的双环芳烃主要为萘类、联苯类及芴类化合物,将1-甲基萘、2-甲基联苯以及芴按不同量定量加入国Ⅵ柴油中,通过对比实验测定多环芳烃含量,首先考察HPLC及GC-MS法进行定量添加双环芳烃(添加量以质量分数计)的准确性。加标样品对应编号:加入2% 1-甲基萘记为加标1;加入2% 2-甲基联苯记为加标2;加入2% 芴记为加标3;同时加入1% 1-甲基萘、1% 2-甲基联苯和1% 芴记为加标4;同时加入2% 1-甲基萘、2% 2-甲基联苯和2% 芴记为加标5。双环芳烃定量加标回收率对比见表4。从表4可以看出,HPLC及GC-MS法测定双环芳烃定量加标时的实验值虽然与理论值接近,但对于添加了芴的加标样品,HPLC法测定双环芳烃的实验值比理论值表现出更明显的偏大现象。其原因是由于芴的折射率比作为标准物的1-甲基萘的折射率更大,而1-甲基萘的折射率又比2-甲基联苯的折射率大,因此在定量加标时,芴的添加量越多,双环芳烃含量的测定值就会比理论值越偏大。这一现象正是由于示差折光检测器对结构响应的不均一性所造成的。

表4 双环芳烃定量加标回收率对比 w,%
为了进一步确定两种方法进行定量加标的准确性,将2% 1-甲基萘、2% 2-甲基联苯及2%芴的混合双环芳烃定量加入柴油11中,此加氢裂化柴油多环芳烃质量分数仅为0.1%。定量加标后,HPLC法和GC-MS法测得双环芳烃质量分数分别为6.5%和6.2%,而两种方法对应的理论值为6.1%。由于加入的3种双环芳烃标准物质中芴的折射率大于作为模型化合物的1-甲基萘的折射率,因此HPLC法测定的双环芳烃含量会比实际值偏大,而GC-MS法测定的双环芳烃含量会与实际值更为接近。说明GC-MS法测定的双环芳烃含量更准确。
2.3.3双环芳烃模型化合物折射率对定量结果的影响上述实验结果均表明,示差折光检测器对结构响应具有选择性,模型化合物的折射率对定量分析的结果有很大影响。为考察双环芳烃响应因子对测定结果的影响,使用芴作为双环芳烃标准物重新绘制工作曲线测定芳烃含量,HPLC法测定结果见表5。从表5可以看出,与使用1-甲基萘时相比,使用较大折射率的芴作为双环芳烃标准物时,测定的双环芳烃含量均会有不同程度的减小。图2为分别以1-甲基萘和芴为模型化合物法的双环芳烃工作曲线对比,两条工作曲线的线性系数均大于0.999,由于芴的折射率大于1-甲基萘的折射率,其工作曲线斜率也相应较大,对相同样品进行校正时就会得到偏小的结果。

表5 采用不同双环芳烃模型化合物时HPLC法分析结果
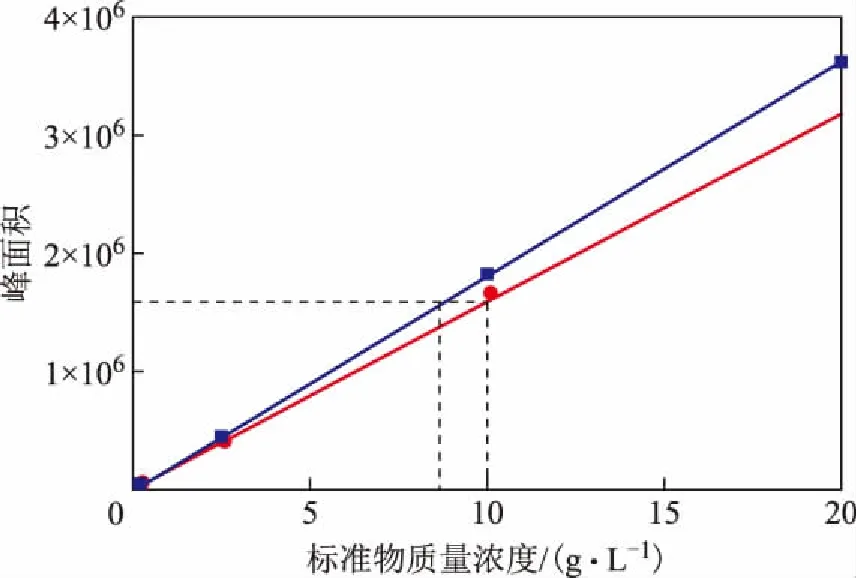
图2 不同双环芳烃模型化合物的工作曲线对比●—1-甲基萘; ■—芴
2.3.4双环芳烃模型化合物相对分子质量对定量结果的影响除了折射率的影响,模型化合物相对分子质量对定量结果也有影响。示差折光检测器是浓度响应型的检测器,其响应信号R可由下式表示:
式中:Z为仪器常数;n0为溶剂的折射率;ni为溶质的折射率;wi为溶质质量分数;Mr为溶质摩尔质量。信号是摩尔浓度响应的,信号响应值与样品组分的相对分子质量成反比。基于此原因,表1中柴油3和柴油4(催化裂化柴油样品)的双环芳烃液相色谱测定值会比质谱法测定值偏大,催化裂化柴油相较于普通直馏柴油或者成品柴油,芳烃侧链会更短,当实际样品组分的相对分子质量小于模型化合物的相对分子质量时,信号响应会偏大,也即测得的芳烃含量偏大。
2.4 国Ⅵ车用柴油对比分析
分别用HPLC和GC-MS法对8个国Ⅵ车用柴油样品中芳烃含量进行对比分析,结果见表6。从表6可以看出,对于这8个国Ⅵ车用柴油,HPLC法测定的单环芳烃和总芳烃含量均比GC-MS法的测定值高,分别平均高3.5百分点和2.7百分点,而HPLC法测定的多环芳烃含量均比GC-MS法测定的多环芳烃含量偏低,平均低0.8百分点。
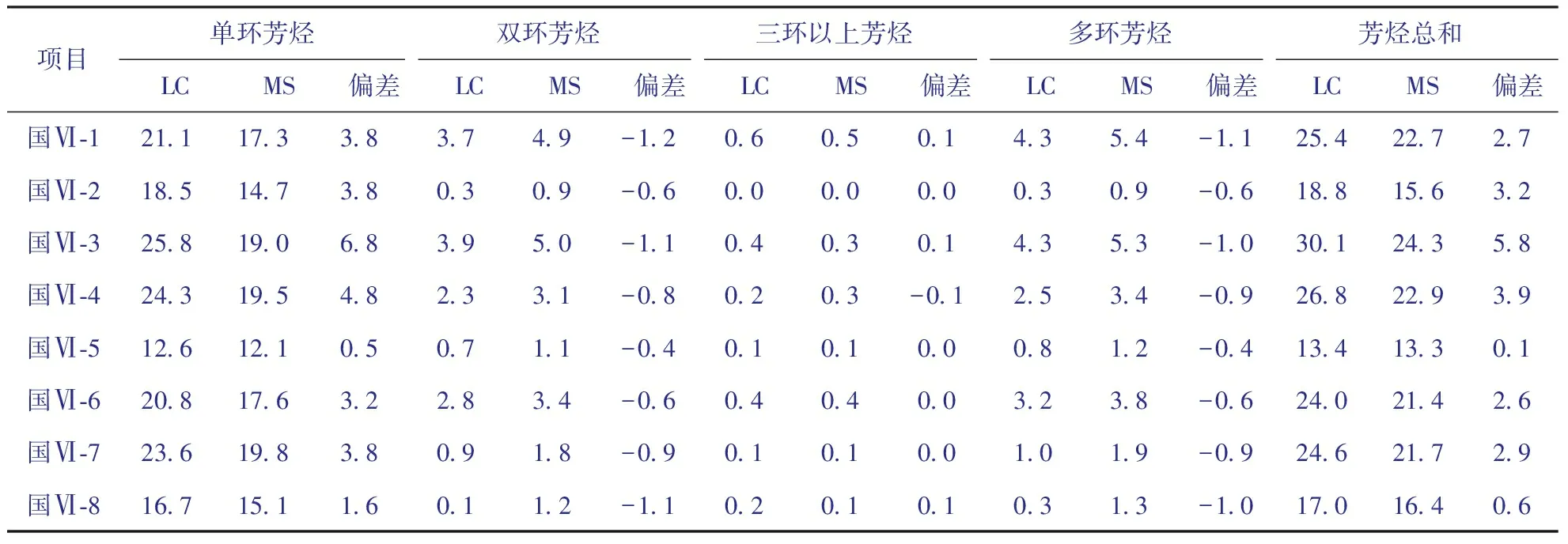
表6 采用HPLC及GC-MS法测定国Ⅵ车用柴油样品中芳烃含量的结果对比 w,%
3 结 论
(1)基于SH/T 0806—2008的HPLC法和基于SH/T 0606—2005的GC-MS法在测定柴油中芳烃含量时,HPLC法测定的总芳烃和单环芳烃含量普遍比GC-MS法偏高;对于大部分直馏柴油和成品柴油,HPLC法测定的多环芳烃含量与GC-MS法相比普遍偏低,但对于少数催化裂化柴油,HPLC法测定的多环芳烃含量相较于GC-MS测定结果则会出现偏高的现象。
(2)HPLC法选用单一模型化合物分别作为单环芳烃、双环芳烃、三环及三环以上芳烃的标准物进行定量,但不同结构的化合物的折射率存在一定的差异。由于柴油分子组成的复杂性,存在大量与模型化合物结构不同的化合物,致使HPLC法在测定单环芳烃、双环芳烃时产生一定的误差,也使由HPLC法得到的多环芳烃和总芳烃含量存在一定的偏差。
(3)掺兑及加标实验结果表明,GC-MS法可提供更准确、详细的柴油芳烃化合物类型分布。
参 考 文 献
[1] 白正伟,李怿,林玉,等.柴油芳烃含量测定方法评价[J].测试与评定,2009(3):86-91
[2] 苟爱仙,李晓刚,苑俊杰,等.柴油中芳烃含量的分析方法探讨[J].炼油与化工,2011,22(4):60-62
[3] Jewell D M,Weber J H,Bunger J W,et al.Ion-exchange,coordination,and adsorption chromatographic separation of heavy-end petroleum distillates[J].Analytical Chemistry,1972,44(8):1391-1395
[4] Mckay J F,Latham D R.High performance liquid chromatographic separation of olefin,saturate,and aromatic hydrocarbons in high-boiling distillates and residues of shale oil[J].Analytical Chemistry,1980,52(11):1618-1621
[5] Suatoni J C,Swab R E.HPLC preparative group-type separation of olefins from synfuels[J].Journal of Chromatographic Science,1980,18(8):375-378
[6] Dark W A.Crude oil hydrocarbon group separation quantitation[J].Journal of Liquid Chromatography,1982,5(9):1645-1652
[8] ASTM D6591-06.Standard Test Method for Determination of Aromatic Hydrocarbon Types in Middle Distillates—High Performance Liquid Chromatography Method with Refractive Index Detection[S].2006
[10] ASTM D2425-04.Standard Test Method for Hydrocarbon Types in Middle Distillates by Mass Spectrometry[S].2004
[11] 张大伟,祝馨怡,田松柏,等.响应因子对高效液相色谱法测定柴油族组成的影响[J].石油与天然气化工,2007,36(2):153-156
猜你喜欢
数学小灵通(1-2年级)(2022年12期)2022-12-23 05:38:18
中国自行车(2022年6期)2022-10-29 01:59:10
食品研究与开发(2022年1期)2022-01-24 11:40:00
水产学杂志(2021年4期)2021-10-18 07:14:30
中国-东盟博览(政经版)(2017年5期)2017-05-08 09:21:19
读写算(上)(2016年4期)2016-12-01 03:19:52
公民与法治(2016年24期)2016-05-17 04:21:53
电源技术(2016年2期)2016-02-27 09:05:12
化工进展(2015年6期)2015-11-13 00:27:25
中国医疗美容(2015年5期)2015-02-03 03:01:43
