
UV法测定益母草总生物碱含量及其不确定度评价
2018-06-05 06:57鄢星魏惠珍吴柳瑾张晓男张丹肖雄饶毅
食品研究与开发 2018年11期
鄢星,魏惠珍,,吴柳瑾,张晓男,张丹,肖雄,饶毅,,*
(1.江西中医药大学药学院,江西南昌330006;2.中药固体制剂制造技术国家工程研究中心,江西南昌330006)
不确定度,依据ISO/IEC指南给出的定义,指表征合理赋予被测量之值分散性,是对测定结果准确性评估的参数,是一种科学、合理的质量控制方法及验证手段[1-3]。与方法学验证一样,是分析学科的重点和难点,2020年版药典,拟制增加《药品检测结果的不确定度评定指导原则》的征求意见稿,因此,随着中国实验室国家认可委员会和国际标准ISO17025对实验室工作规范要求的开展,建立测量不确定度评价分析方法将成为药学分析领域的必要步骤[4-5]。
益母草是唇形科益母草属植物地上部分及,主治妇女月经不调,胎漏难产,产后血晕等病症,是治疗子宫疾病常见的中药[6]。生物碱是益母草的主要化学成分,具有显著的抗炎、镇痛等药效活性[7-8]。有文献研究报道[9-10],生物碱含量与益母草的作用功效之间存在正相关的量效关系,认为总生物碱含量越高,益母草的药理药效作用越明显。因此,准确测定益母草中的总生物碱的含量,对整体控制益母草药材质量具有重要现实意义。
本文采用了紫外分光光度法测定益母草中总生物碱含量,对10批益母草中总生物碱含量测定结果进行不确定度评估,保证分析方法的可行性和合理性,同时确保了试验结果的准确性和有效性,为使用紫外分光光度仪测定中药有效成分含量的不确定度研究提供参考。
1 材料与方法
1.1 试剂与仪器
益母草分别采自平邑、四川、广西、贵州、河南5个产地10批样品,经江西中医药大学药学院植物鉴定学老师张中立检定为唇形科植物益母草(Herba Leonuri Houtt),样品信息见表1。
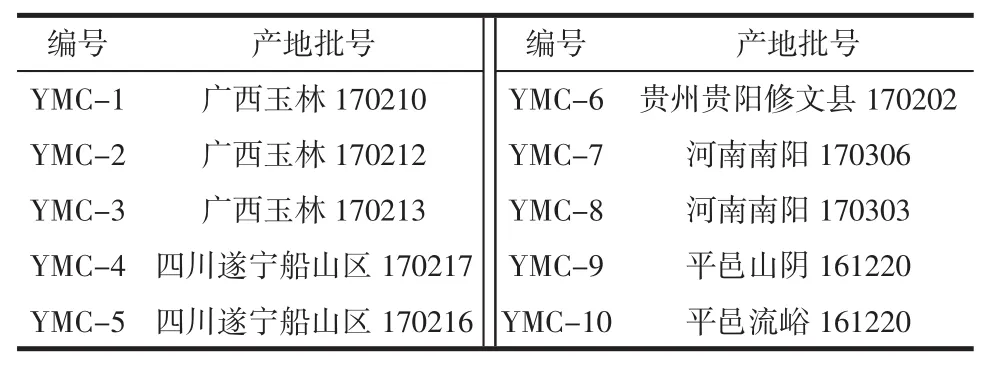
表1 益母草信息Table1 Information on Herba Leonuri
雷氏盐(分析纯):国药集团化学试剂有限公司;盐酸(优级纯):西陇化工股份有限公司;无水乙醇、95%乙醇(分析纯):上海振兴化工一厂;盐酸水苏碱对照品(批号S110214,规格1 g,含量以98%计):上海阿拉丁生化科技股份有限公司;Milipore超纯水。
UV-2550紫外可见分光光度仪、AUW2200十万分之一天平:日本岛津公司;KQ-250DB型数控超声波清洗器:昆山市超声仪器有限公司;AB104-N万分之一天平:梅特勒-托利多公司;Mlli-Q超纯水仪:美国Milipore公司。
1.2 方法
1.2.1 标准曲线绘制
精密称取盐酸水苏碱对照品30.52 mg,加入0.1 mol/L 的盐酸溶液定容为50 mL 吸取 2、5、8、10、15 mL标准溶液于25 mL容量瓶中,加入新配置的2%雷氏盐溶液(临用现配)6 mL,用0.1 mol/L的盐酸溶液定容,摇匀。置冰浴1 h,干燥滤纸过滤,取续滤液作为标准品溶液。取6 mL新配制的2%雷氏盐溶液至25 mL容量瓶中,加0.1 mol/L盐酸溶液定容作为空白溶液。在530 nm测定标准溶液吸收值(Ai)和空白溶液的吸收值A空白,以y=A空白-Ai值为纵坐标,以标准溶液中总生物碱的浓度x(mg/mL)为横坐标,计算标准曲线 y=1.840 1x-0.138 3,r=0.999 2。
1.2.2 供试品溶液的配制
精密称取益母草药材粉末(过3号筛)约5.0 g,加入0.1 mol/L的盐酸溶液100 mL,超声30min,提取2次,合并滤液,水浴蒸至流浸膏,用0.1 mol/L盐酸溶液多次洗涤溶解至50 mL容量瓶中,取10 mL供试品溶液,加5 mL 2%雷氏盐溶液,用0.1%盐酸溶液定容为25 mL,置冰浴1 h,干燥滤纸过滤,取续滤液,按照“1.2.1”标准曲线的方法进行样品测定。
2 样品测定结果
按照2.2制备供试品溶液,在530 nm处测定样品溶液的吸收值As,以空白溶液调零,将A=A空白-As值代入标准曲线方程中计算总生物碱含量,见表2。

表2 10批益母草药材总生物碱含量测定结果(n=2)Table 2 Determination of total alkaloid content of 10 batches of Herba Leonuri(n=2)
3 不确定度评价分析
3.1 不确定度来源分析
依据上述建立的模型及试验方法可知,总生物碱含量测定的不确定来源主要有以下几个方面:1)标准品引入的不确定度Urel(i);2)拟合线性标准曲线的不确定度Urel(Yi);3)供试品引入的不确定度Urel(s);4)仪器测定的不确定度Urel(A);5)样品重复测定不确定度Urel(F)。各分量之间关系结构如图1。
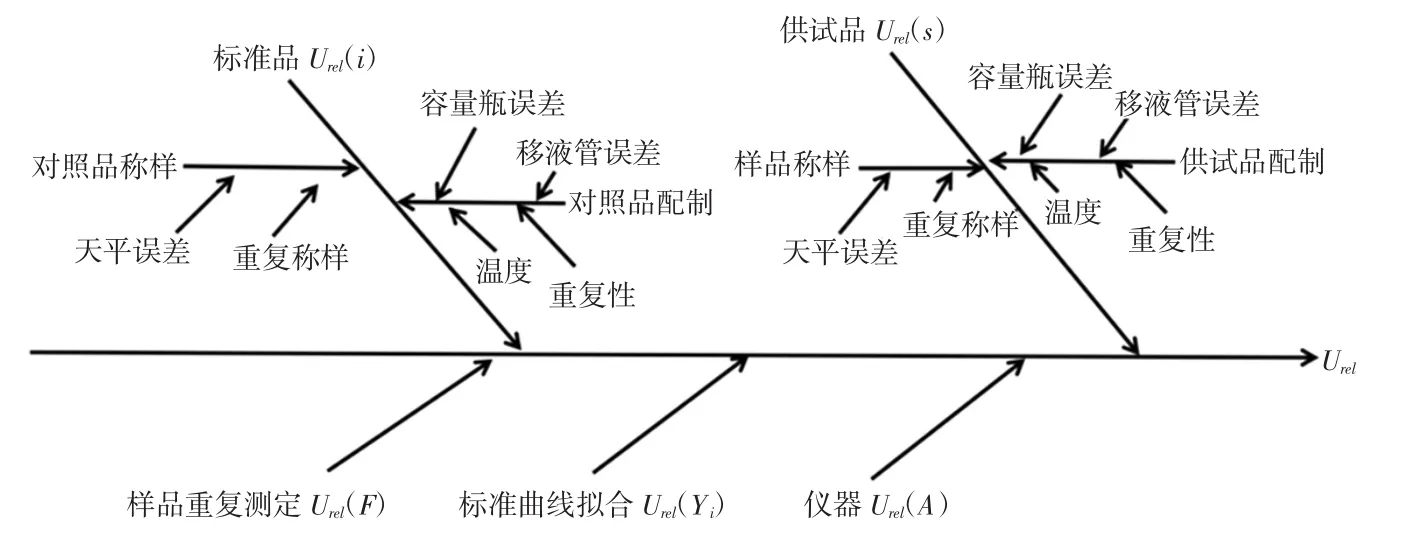
图1 各分量不确定度关系结构图Fig.1 The structure diagram of the relation of the uncertainty of each component
3.2 各分量的不确定度分析
依据《测量不确定度评定与表示》的指示,对于明确区间范围但未给出误差置信水平的,如标准品纯度、天平示值和温度变化的影响按矩形分布取值k=;JJG196-2006《玻璃量器检定规程》[11]给出了移液管、容量瓶等量器的误差区间,没有说明置信水平,参照相关文献[12-13],采用三角分布取值计算不确定度;对于容量瓶、仪器重复性测定引入的不确定度,认为服从正态分布,根据试验结果的标准偏差计算不确定度。
3.2.1 标准品引入的不确定度
3.2.1.1 标准品称样
标准品称样引入的不确定度的来源有天平的示值误差和重复称样的误差。查阅十万分之一天平检定报告可知,其允许误差为±0.1 mg,依据矩形分布取值,则天平示值误差引入的标准不确定度u1(mi)=。由于天平检定报告证书没有给出最大允许误差,通过重复称量样品(n=6),计算天平重复测量带来的标准偏差为0.3 mg,服从正态分布,则天平重复称量可达到的最大标准不确定度。标准品的称样量为30.52 mg,综上可得,标准品称样引入的相对标准不确定度。
3.2.1.2 标准品溶液配制
标准品在配制过程中用到了的玻璃量具有15 mL单标移液管,10 mL分标移液管,25、50 mL的容量瓶,均属A类玻璃量具,其不确定度来源有3个:玻璃量器的校准、重复测量和温度变化。
根据JJG196-2006《常用玻璃量器检定规程》,10 mL分标、15 mL单标移液管和25、50 mL容量瓶的允许误差分别为±0.05、±0.025、±0.03、±0.05 mL,通过对以上玻璃量器的重复验证(n=6)试验得到的重复测量带来的标准偏差分别为0.08、0.05、0.05、0.07 mL,此外,室温对定容和量取的体积有影响,其量取和校准最适温度在20℃,最大允许温度误差为±5℃,水的膨胀系数为N=2.1×10-4/℃,则标准品溶液配制过程中各分量不确定度计算结果见表3。

表3 标准品溶液配制引入的不确定度Table 3 Uncertainty in the introduction of standard solution preparation

续表3 标准品溶液配制引入的不确定度Continue table 3 Uncertainty in the introduction of standard solution preparation
3.2.2 样品引入的不确定度
3.2.2.1 样品称样
样品称样使用的是万分之一天平,根据天平检定报告证书可知,允许误差为±1 mg,按矩形分布取值,则天平示值引入的不确定度0.577 3。由于天平检定报告证书没有给出最大允许误差,通过重复称量样品(n=6),计算天平重复测量带来的标准偏差为1.5 mg,服从正态分布,则天平重复测量引入的不确定度。
综上,供试品称样引入的标准不确定度u(ms)=,15批供试品的平均称样量ms为5 004.1 mg,则相对标准不确定度0.000 2。
3.2.2.2 样品溶液配制
供试品在配制过程中用到的玻璃量具有10、100 mL单标移液管和25、50 mL容量瓶,均属A级玻璃量具,根据JJG196-2006《常用玻璃量器检定规程》,其中10、100 mL单标移液管的允许误差分别为±0.02、±0.08 mL,重复测量带来的标准偏差分别为0.05、0.11 mL,其他同“3.2.1.2”。供试品溶液配制的各分量不确定度计算结果见表4。
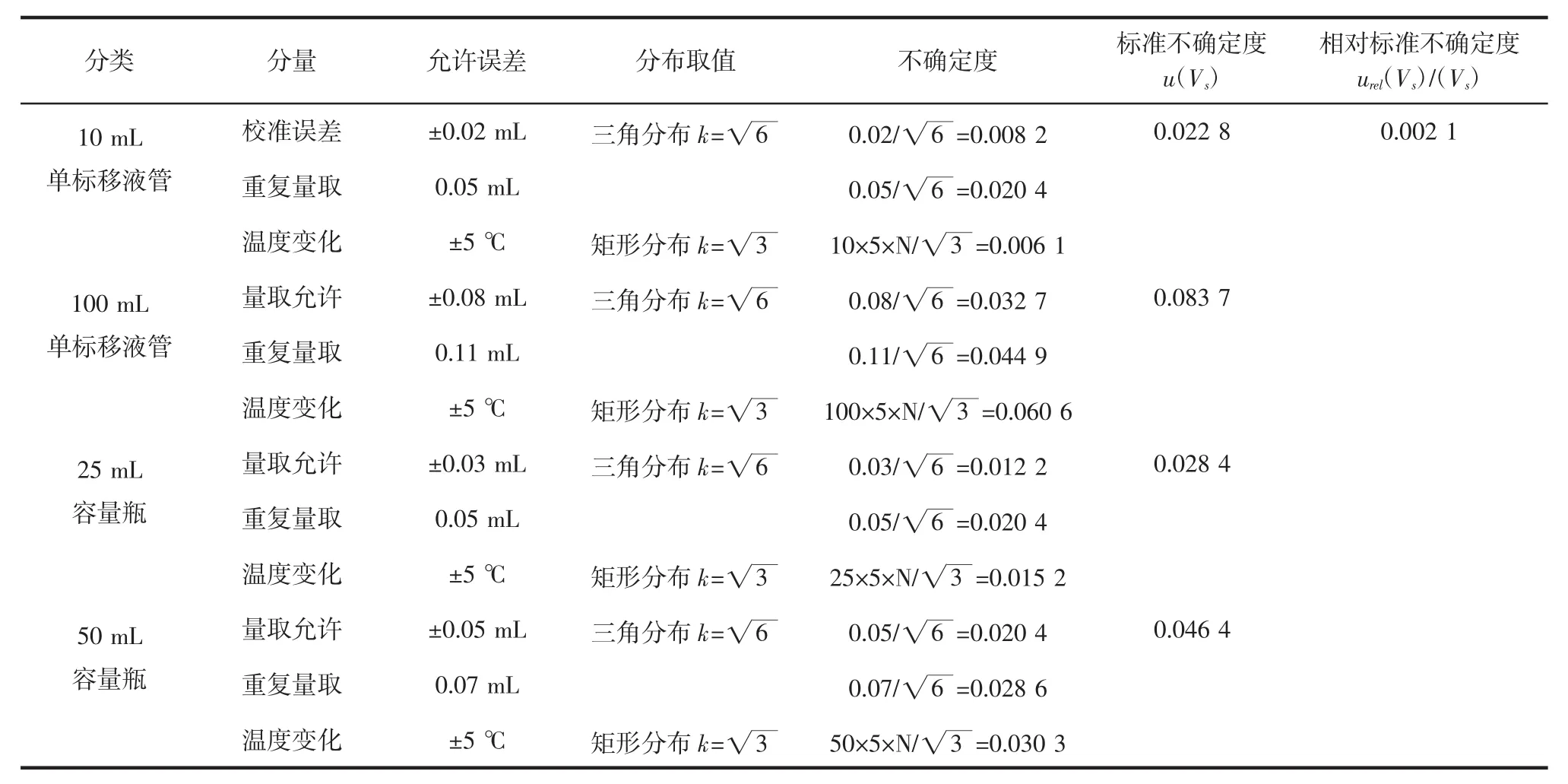
表4 供试品溶液配制引入的不确定度Table 4 Uncertainty in the introduction of the preparation of the sample solution
3.2.3 标准曲线拟合引入的不确定度
本试验配制5个不同溶度的标准溶液(浓度由低到高):0.120 1、0.295 0、0.449 9、0.774 8、1.039 9 mg/mL,每个浓度的平均吸光值分别为0.1358、0.2411、0.317 8、0.502 8、0.635 0(每个浓度测 2 次),根据赛贝尔公式,计算标准曲线方程的标准偏差Si,则由拟合标准曲线求得供试品Cs引入的标准不确定度u(Yi)和相对标准不确定度urel(Yi)计算如下:
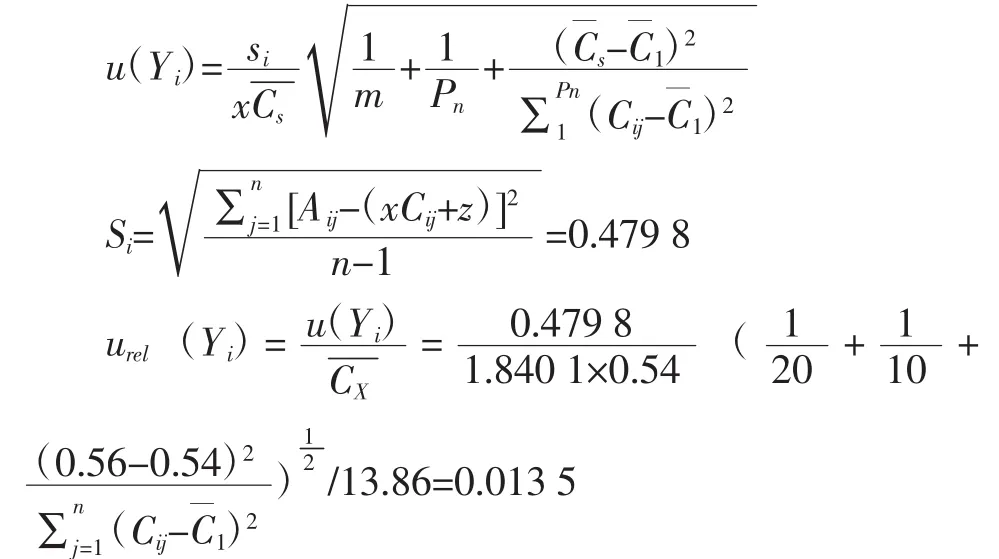
式中:j标准溶液的测定顺序(浓度由高到低j=1,2…n);Aij为对应顺序 j标准品溶液的吸光值;Cij为对应顺序的标准品溶液的浓度;x为标准曲线系数(x=1.840 1);z为标准曲线的截距(z=-0.138 3);s为10批供试品溶液的平均浓度(由表2可计算出s=0.56 mg/mL);m为供试品溶液测定的次数(10批样品,每批平行测定2次,m=20);Pn为标准品溶液测定的次数,(Pn=10);1为标准品溶液的平均浓度(1=0.54mg/g);为10批样品的平均含量(由表2可计算出13.86 mg/g)。
3.2.4 仪器引入的不确定度
主要由仪器本身误差和仪器重现性测定两个方面引入的不确定度。该紫外分光光度仪的检定报告证书指出了测量结果的准确性误差为±0.002,按矩形分布,标准不确定度。对10份样品测定吸光值,求得10份样品平均浓度为0.236 1 mg/mL,根据赛贝尔公式计算标准偏差为0.019 5。假设服从正态分布,仪器重现性引入的标准不确定度。则仪器引入的相对标准不确定度:
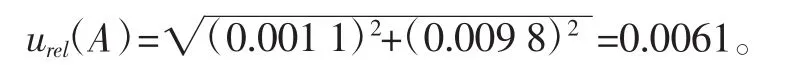
3.2.5 样品重复测定引入的不确定度
根据“2.4.3”同一份样重复测定,6 次测得的含量分 别 为6.078 4、5.891 3、5.789 6、6.000 7、5.865 5、5.936 3 mg/g,平均结果为5.926 9 mg/g,根据赛贝尔公式计算含量测定结果的标准偏差SF=0.009 87,服从正态分布考虑,样品重复测定的标准不确定度,相对标准不确定度
3.3 合成、扩展与报告不确定度的计算
由已计算出各分量的相对标准不确定度,合成相对标准不确定度可由各分量的相对标准不确定度按下式合成:。

由此得到合成标准不确定度 U(CX)=Urel×CX[CX为每批样品中总生物碱平均含量,(mg/g),见表2;X为每批样品的编号];不确定度贡献率计算公式为M/%=[urel(W)]2/[Urel]2×100(W为表5中各个不确定度分量)。各分量的相对标准不确定度及贡献率统计结果见表5。取置信水平为95%,包含因子k=2,则扩展不确定度为U(X)=U(CX)×k,由“2.5”试验结果可知,10 批益母草中总生物碱含量在 19.09 mg/g~2.47 mg/g,报告不确定度表示为CX±U(X),结果见表6。
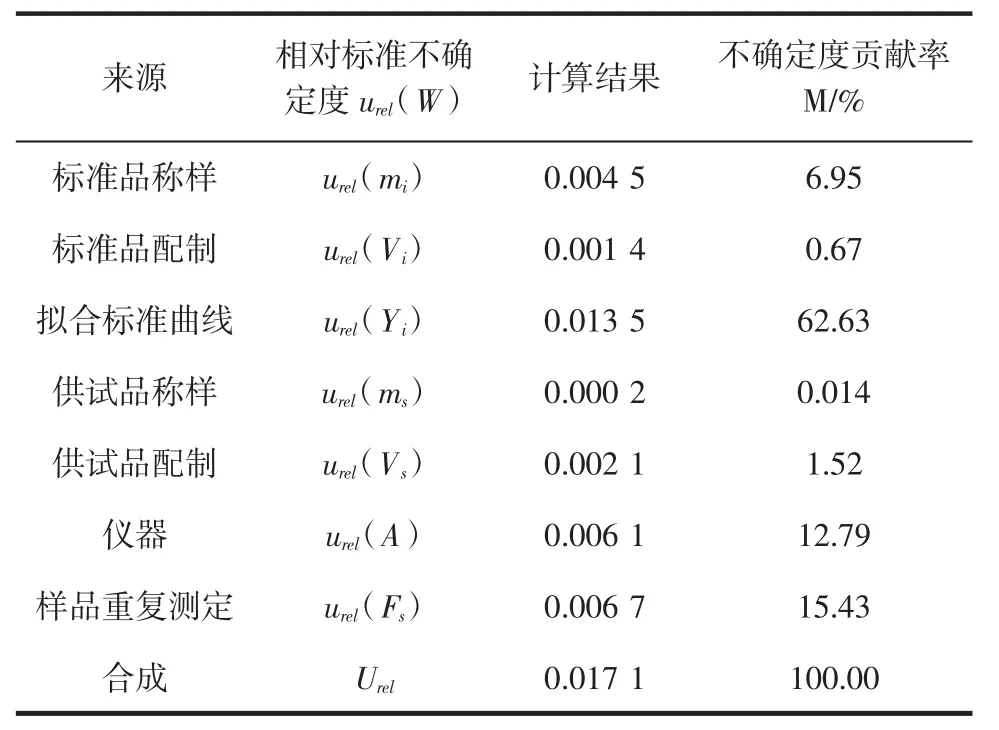
表5 各分量不确定度计算结果及贡献率Table 5 Calculation results and contribution rate of each component uncertainty
4 讨论与小结
雷氏盐在酸性介质中可与绝大部分生物碱反应,定量的生成不溶于水的雷氏铵盐沉淀[14],常规测定方法是将沉淀溶解于丙酮中,直接测定溶液的紫外吸光度,根据Beer-Lambert公式计算含量。由于沉淀的溶解较为繁琐,丙酮溶剂本身对人体有害,故本试验在常规方法上进行改进,通过测定滤液中剩余雷氏盐的吸光度[15],以空白溶液吸光度减去滤液的吸光度,间接测得生物碱含量,改进后的方法操作简单,重现性好。
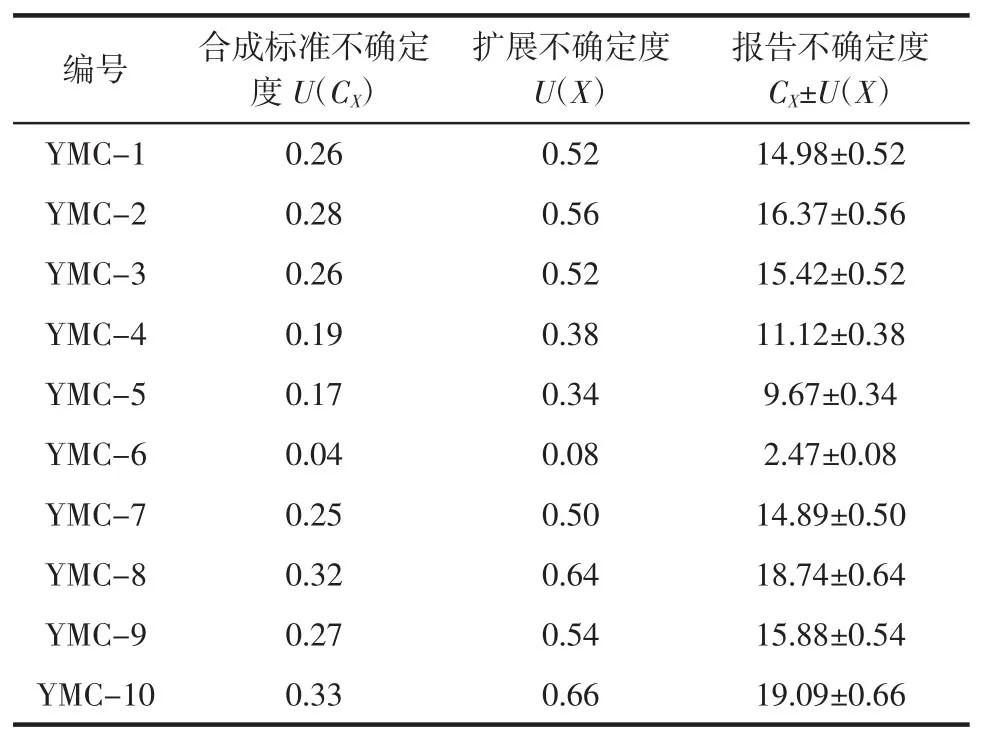
表6 总生物碱合成不确定度、扩展不确定度与报告不确定度计算结果Table 6 Calculation results of total alkaloids of synthetic uncertainty,expansion uncertainty and report uncertainty mg/g
本试验运用不确定度理论对紫外分光光度法测定益母草总生物碱含量过程进行评定研究,从表5各分量的标准不确定度计算结果可知,拟合标准曲线和样品重复测定是不确定度的主要来源,拟合标准曲线的合成贡献度最大为62.63%,说明本方法测定结果的不确定度主要由拟合标准曲线引入的,由拟合标准曲线不确定度计算过程可知,可能是标曲建立选取的浓度点过少,以及检测样品的含量差异过大引起的,建议标准曲线建立的浓度点增加至5个以上,拉大线性范围,使所有浓度的样品在线性范围之内;其次是样品重复测定带来的不确定度,贡献度为15.43%,在实际检测样品应严格样品制样引入的不确定度,规范实验操作,减少人为因素对样品重复测定过程中的影响。同时,为了简化分析过程,本试验忽略了提取过程中超声仪和沉淀显色剂雷氏盐带来的不确定度影响。
目前,有关紫外分光光度法对中药中有效成分定量测定方法的不确定度分析较少,2015版《中国药典》规定了对所建立的方法进行方法学验证,但未对试验具体结果作定量的置信性分析,本文以益母草总生物碱含量测定为例,建立紫外光光度法测定中药中有效成分的质量控制方法,并对该方法在试验过程中给测定结果带来的不确定度进行了定量分析,保障了测定方法的可行性、合理性和试验结果的准确性、有效性,降低了该方法在日常应用中的风险,同时为其他分析方法测定中药有效成分的不确定度定量评价提供参考。
[1]ISO/CASCO,ISO/DIS 17025:2017.General requirements for the competence of calibration and testing laboratories[EB/OL].[2018-01-30].https://www.iso.org/standard/39883.html
[2]国家质量监督检验检疫总局.JJF1059.1-2012测量不确定度评定与表示[S].北京:中国标准出版社,2013:16
[3]王馨,徐冰,薛忠,等.中药陈皮提取物粉末中糊精含量近红外分析方法的验证和不确定度评估[J].药物分析杂志,2017,37(2):339-344
[4]Ellison S L R,Rosslein M,Williams A.Quantifying Uncertainty in Analytical Measurement[M].2nd Ed.UK:Teddington EURACHEM/CITAC,2000:11-13
[5]中国实验室国家认可委员会.化学分析中不确定度的评估指南[M].北京:中国计量出版社,2002:11
[6]孙蓉,冯群,赵庆华,等.益母草毒性研究进展[J].中国药物警戒,2014,11(2):70-73
[7]Miao M,Tian S,Ming B,et al.Effect of Motherwort total alkaloids on the prostate hyperplasia mice model of pathological changes of related tissue morphology induced by the fetal urogenital sinus implants[J].Saudi Pharmaceutical Journal,2017,25(4):601-606
[8]赵红,李世民,窦立雯,等.益母草生物碱药理毒理学研究进展[J].中国药物警戒,2015,12(12):722-726
[9]何丽,杨俊毅,张静,等.益母草注射液与缩宫素联合应用预防产后出血的临床疗效[J].中国妇幼保健,2010,25(11):1571-1573
[10]于长莉,王昊珏.应用益母草注射液联合缩宫素预防产后出血的临床分析[J].中国计划生育和妇产科,2012,4(2):56-58
[11]国家质量监督检验检疫总局.JJG 196-2006常用玻璃量器检定规程[S].北京:中国计量出版社,2006
[12]BLEYE CD,CHAVEZ PF,MANTANUS J,et al.Critical review of near-infrared spectroscopic methods validations in pharmaceutical applications[J].J Pharm Biomed Anal,2012,69:125-132
[13]CARDENAS V,CORDOBES M,BLANCO M,et al.Strategy for design NIR calibration sets based on process spectrum and model space:An innovative approach for process analytical technology[J].J Pharm Biomed Anal,2015,114:28-33
[14]袁王俊,张维瑞,李勉,等.不同产地益母草中总生物碱含量的比较[J].河南大学学报(医学版),2013,32(1):38-39
[15]许小华,杜绍亮,杨云.益母草中总生物碱的提取及含量测定[J].河南科学,2008,26(12):1481-1482
猜你喜欢
中草药(2022年1期)2022-01-13
中学生数理化·八年级物理人教版(2021年12期)2021-12-31
中学生数理化·八年级物理人教版(2020年12期)2021-01-18
中成药(2018年11期)2018-11-24
少年漫画(艺术创想)(2018年2期)2018-09-11
中成药(2017年12期)2018-01-19
中成药(2017年7期)2017-11-22
中成药(2017年8期)2017-11-22
中成药(2017年5期)2017-06-13
中国当代医药(2015年24期)2015-03-01
