
原发性侧索硬化临床及影像特征分析
2018-05-22 03:13刁东卫刘建国夏德雨戚晓昆
大连医科大学学报 2018年2期
刁东卫,刘建国,夏德雨,戚晓昆
(1. 大连医科大学 研究生院,辽宁 大连116044;2. 海军总医院 神经内科,北京100048)
原发性侧索硬化(primary lateral sclerosis,PLS)是一种非常少见的运动神经元病,病因及发病机制仍不明确,临床表现为进行性上运动神经元功能障碍。多在中年以后发病,起病隐袭,进展慢,可存活较长时间。PLS非常少见,大约占所有运动神经元病患者的1%~4%,国内仅有少数文献报道。本文通过分析海军总医院神经内科收治的4例PLS患者临床资料,并复习相关文献,讨论其临床、影像学表现及鉴别诊断。
1 临床资料
病例1:患者男性,69岁,主因“右侧肢体活动笨拙5年”入院。患者2012年无明显诱因逐渐出现右下肢笨拙,当地诊断“腰椎间盘突出”,给予针灸等治疗无好转。2016年7月出现右上肢笨拙,言语不清,右下肢疼痛,并反复摔倒多次。2016年10月需拐杖辅助行走,蹲起困难。病程中无饮水呛咳及吞咽困难,2017年3月就诊于海军总医院。既往体健,家族中无类似疾病史。神经系统查体:轻度构音障碍,痉挛步态,四肢肌容量正常,左侧肢体肌力5级,右侧肢体肌力5-级,双下肢肌张力高,右侧更显著,四肢腱反射亢进,双侧Hoffmann征阳性,双侧Babinski征阳性,双侧踝阵挛阳性。辅助检查:血常规、血生化、血沉、C-反应蛋白、免疫四项正常,甲状腺功能七项、血清狼疮组套、风湿组套、肿瘤标志物均正常,脑脊液压力、常规、生化、OB、MBP、IgG合成率均未见异常。肌电图:四肢运动感觉神经传导速度正常,针极肌电图EMG:运动单位电压(μV)第1骨间肌1753↑、右股四头肌1426↑,时限均正常,余脊旁肌、胸锁乳突肌肌电图均未见异常。头颅MRI:未见明显异常。颈椎MRI:C4-5椎间盘轻度膨出,颈髓未见明显受压。胸椎MRI:大致正常。入院给予巴氯芬缓解肌张力及丁苯肽、维生素B1、甲钴胺、辅酶Q10等治疗,4个月后随访,患者症状无明显加重。
病例2:患者男性,48岁,主因双下肢无力8个月入院,2012年3月无明显诱因出现双下肢无力,2012年10月症状加重、走路不稳,就诊于当地医院考虑脱髓鞘病,给予甲强龙冲击治疗,症状无明显好转,2012年11月就诊于海军总医院。既往体健,无家族史。查体:下颌反射阳性,肌容量正常,双下肢肌力4+级,四肢腱反射亢进,双侧Hoffmann征阳性,双侧Babinski征阳性,双侧Chaddock征阳性,双侧踝阵挛阳性。血常规、血生化、血沉、C-反应蛋白正常,肿瘤标志物、免疫四项未见异常,脑脊液常规、生化正常,血清及脑脊液OB、MBP、AQP-4抗体均阴性,肌电图:四肢感觉运动神经传导速度正常,胸锁乳突肌、脊旁肌、左肱二头肌、左股四头肌针极肌电图运动单位电压及时限均未见特征性改变。头颅MRI:双侧皮质脊髓束走行区可见对称长T2信号(图1),颈椎MRI未见明显异常。入院后给予丁苯肽、维生素B1、甲钴胺、辅酶Q10治疗10 d,病情无明显变化,出院后继续口服上述药物,2017年5月(5年后)随访,患者双下肢无力较为缓慢加重,需拄拐杖行走。
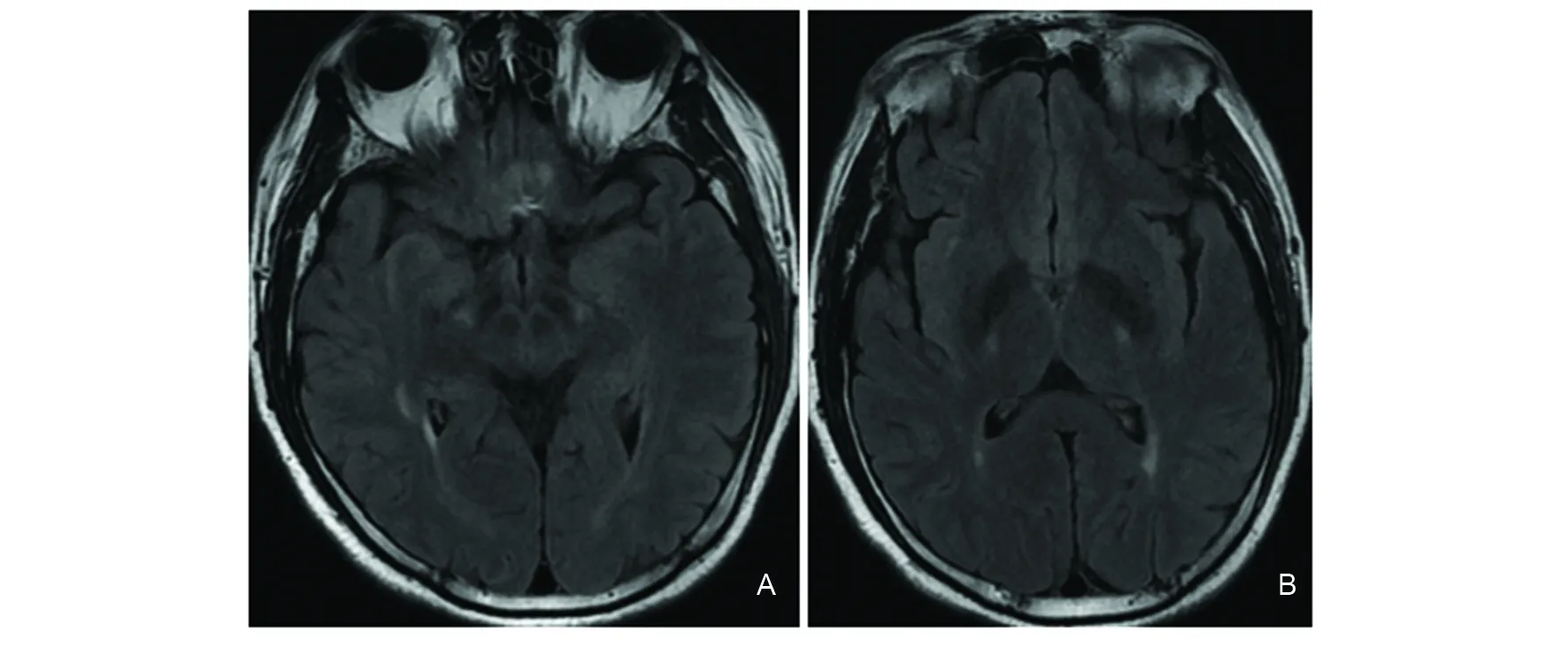
A:T2Flair双侧大脑脚对称高信号;B:双侧内囊后肢对称高信号图1 病例2头颅MRIFig 1 Head MRI of case 2
病例3:患者男性,39岁,因“缓慢进行性双下肢无力6年”入院。患者2009年无明显诱因出现双下肢无力,右侧重于左侧,尚能行走,2012年走路拖拽,伴饮水呛咳,言语缓慢。2015年不能独立行走,就诊于海军总医院。既往体健,家族中无遗传病史。查体:言语缓慢,肌容量正常,左下肢肌力4级,右下肢肌力3级,双下肢肌张力增高,四肢腱反射亢进,双侧Hoffmann征阳性,双侧Babinski征、Chaddock征阳性。血常规、血生化、免疫四项、肿瘤标志物筛查未见异常,肌电图:四肢感觉运动神经传导速度正常,针极肌电图(EMG)示运动单位轻收缩时限(ms):左股四头肌13.7↑(24%),右股四头肌正常,电压(μV):左股四头肌1657↑,右股四头肌1388↑,肛门括约肌肌电图未见特征性改变。头颅MRI:桥脑、双侧大脑脚、内囊后肢皮质脊髓束走行区可见对称性长T2信号,左侧略重于右侧(图2)。颈胸椎MRI:颈胸段脊髓萎缩变细。入院给予丁苯肽、维生素B1、甲钴胺、辅酶Q10治疗17 d,病情未见明显好转,出院后继续口服上述药物。2年后随访,患者不能行走,双手笨拙。
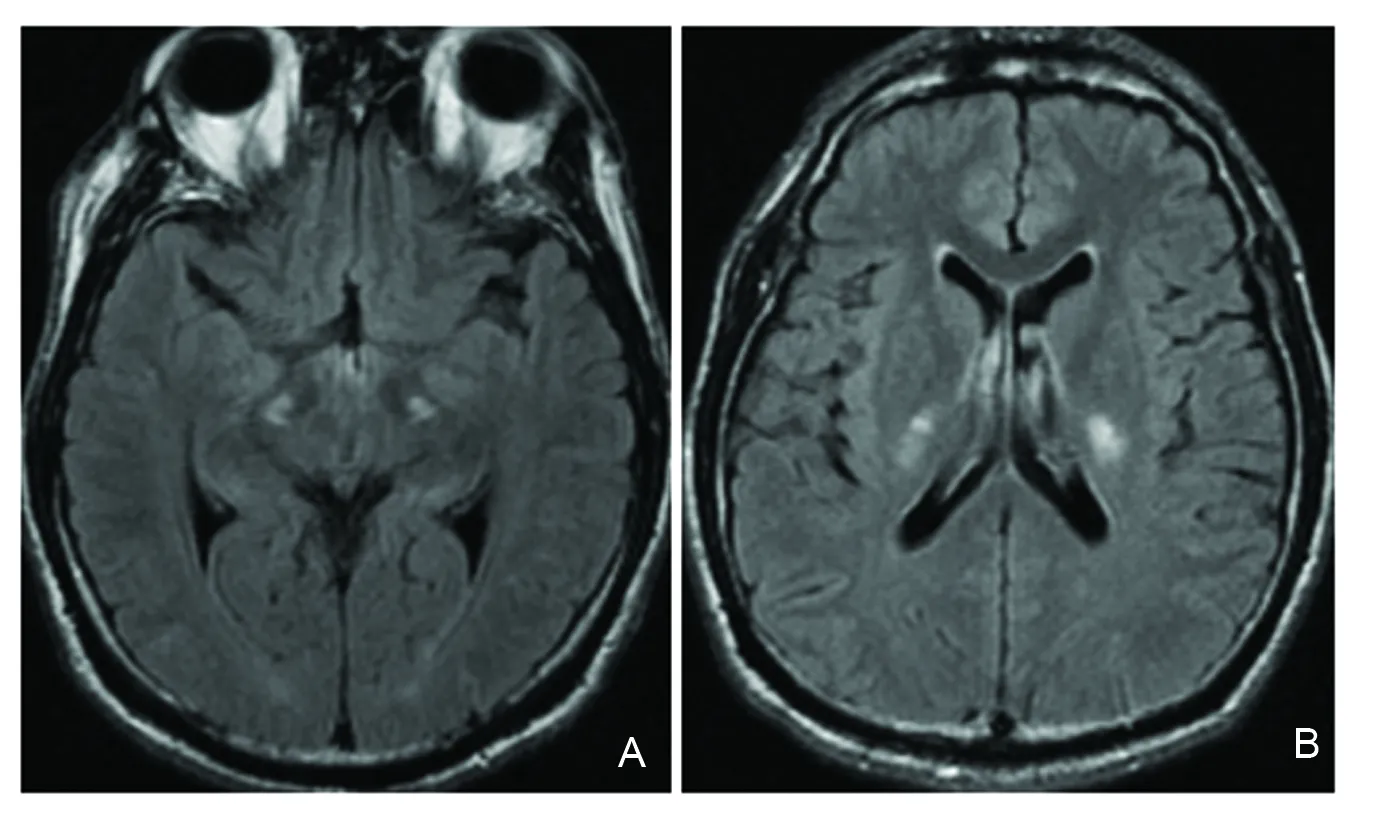
A:T2Flair双侧大脑脚对称高信号;B:双侧内囊后肢对称高信号图2 病例3头颅MRIFig 2 Head MRI of case 3
病例4:患者女性,64岁,主因面部多汗2年,双下肢无力8个月入院。患者2012年无明显诱因出现面部多汗,无尿便障碍,无睡眠障碍,未予治疗,8个月前出现双下肢无力,不能长久站立,行走不稳,当地医院诊断“多系统萎缩”,建议外院诊治,2014年8月就诊于海军总医院。既往体健,无家族史。查体:四肢肌容量正常,双下肢肌力5-级,双下肢肌张力高,四肢腱反射亢进,双侧Hoffmann征阴性,双侧Babinski征、Chaddock征阳性。血常规、血生化、肿瘤标志物、免疫四项未见异常,脑脊液常规、生化正常。肌电图:四肢神经传导速度、针极肌电图未见异常,SSR左右手、左右足潜伏期在正常范围,肛门括约肌运动单位电压时限未见特征性改变。头颅MRI正常,颈椎MRI:颈髓稍细。智力评定:MMSE:29分,MoCA:29分。给予口服丁苯肽、维生素B1、甲钴胺、辅酶Q10治疗,该患者随访3年未发现明显自主神经、小脑及锥体外系受累的证据,排除其他诊断,考虑PLS。
2 讨 论
运动神经元病(motor neuron disease,MND)是以上、下运动神经元改变为特征的慢性进行性神经系统变性疾病。临床可表现为肌无力、肌萎缩、延髓麻痹、锥体束征。病因与发病机制目前仍不明确。Strickland等[1]发现,MND与从事焊接有很强的相关性,其它危险职业还包括油漆或颜料制作行业、造船业、电镀、乳制品行业。MND可分为4种类型:肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)、进行性脊肌萎缩(progressive muscular atrophy,PMA)、进行性延髓麻痹(progressive bulbar palsy,PBP)、原发性侧索硬化(primary lateral sclerosis,PLS)。其中以PLS预后最好,但较为少见。PLS是以脊髓、延髓麻痹为特征的上运动神经元变性,病理表现为皮质脊髓束显著的脱髓鞘改变。1992年Pringle等[2]提出PLS的诊断标准:成年隐匿起病的痉挛性麻痹,通常起于下肢,无家族史,至少3年缓慢进展的过程,最终导致严重的痉挛性脊髓麻痹、假性球麻痹和MRI显示大脑中央前回萎缩。随着更多PLS的病例报道,有学者认为PLS可以向ALS转化[3],Wais等[4]对76名PLS患者进了临床和电生理随访研究,发现60%的患者在发病8年后有下运动神经元(lower motor neuron,LMN)受累,认为PLS应该作为ALS的一个亚型。因此,PLS的诊断标准由最初推荐的3年未累及LMN的时间窗延长至4年[5]。至于PLS是否为ALS的亚型还是独立的疾病,目前仍存在争议。根据目前国内外文献报道,虽然二者均有运动障碍,但与ALS相比,PLS疾病发展明显较缓慢、病程长,且预后明显较好,因此我们认为PLS与ALS存在显著差异。
PLS临床表现多样,症状通常首发于50~60岁,最常见的临床症状是脊髓痉挛性麻痹,包括肢体强直、反射亢进、轻度无力。病人主诉包括僵硬、笨拙、不协调,球部症状可以有言语含糊、吞咽困难,1/2~1/3患者有尿频或尿急,认知功能一般不受影响,但也有作者报道PLS有认知功能损害,包括执行功能、记忆和语言受损[6]。少数文章还提及LMN受损,Bruyn等[7]发现10例PLS患者中有1例出现了第1骨间肌、小指展肌萎缩,感觉是正常的,没有束颤。本文4例患者主要表现为下肢无力,随着病情进展2例患者出现上肢笨拙,其中第1、第3例患者有轻度假性球麻痹的症状。查体均有四肢腱反射亢进,双侧病理征阳性。但4例患者均未发现肌萎缩和认知功能障碍。第1例和第3例患者就诊时病程已超过4年,无LMN受累的症状和体征,但例1肌电图显示第1骨间肌、右股四头肌电压增高,病例3患者肌电图示双侧股四头肌运动单位电压增高、左侧股四头肌运动单位时限增宽,这与文献报道的15%PLS有轻度的肌电图改变一致[8]。例2患者随访5年,例4患者随访3年均未发现LMN受累的证据。4例患者临床症状与体征均提示上运动神经元病变,颈髓、胸髓核磁髓内未见明显异常信号,无家族史,结合实验室检查,排除血管性、感染性、肿瘤性、代谢性、炎症性、遗传性病变,考虑变性病,诊断原发性侧索硬化(PLS)。但随着病程的延长,这些患者是否会出现肌萎缩,是否会转化为ALS,仍需继续随访。PLS非常罕见,症状无特异性,临床医生如未仔细询问病史及行细致的神经系统查体,容易导致误诊。第1例患者曾在当地被误诊为“腰椎间盘突出症”,腰椎间盘突出症临床上以持续腰背部钝痛多见,80%以上有下肢放射痛,5%患者有肢体麻木,而因腰椎间盘突出造成瘫痪者十分罕见,该患者无腰痛及感觉障碍,下肢肌张力异常增高,不支持该病。第2例患者被误诊为“脱髓鞘病”,但颈椎MRI未见异常,血清及脑脊液免疫学相关检查均阴性,应用大剂量激素冲击治疗未见明显改善,故可排除脱髓鞘病变。第4例患者因有自主神经和锥体束的症状而被误诊为“多系统萎缩”,但该患者无尿便及睡眠障碍,头颅MRI未发现“壳核裂隙”及桥脑“十字征”等改变,入院后完善肛门、尿道括约肌肌电图未见特征性改变,目前仍未发现小脑及锥体外系受累的证据,应继续进行随访观察。
目前的研究表明,MND可出现相应的颅内病变。Thorpe 等[9]研究发现MND组患者头颅MRI皮质、半卵圆中心、内囊和大脑脚的高信号显著高于正常对照组,Kiernan等[10]报道MND患者头颅MRI可显示前额叶的萎缩,大部分位于中央前回,伴有皮层下白质变性,认为常规MRI可以作为MND的诊断手段,但尚不足以区分其亚型。本文两例患者头颅MRI发现皮质脊髓束走行区对称性T2高信号,符合运动神经元病的诊断。由于PLS与早期以上运动神经元(upper motor neuron,UMN)为主的ALS难以鉴别,近些年学者们热衷于PLS支持性生物标志物的研究,Agosta等[11]的研究发现,PLS比ALS患者DTI显示更严重的皮质脊髓束和经胼胝体运动纤维受累。Iwata等[12]研究发现PLS中FA的减少主要在近皮层的白质,提示锥体细胞变性,其扩散特性改变累及颅内皮质脊髓束的全长。Kuipers-Upmeijer等[13]报道因PLS患者病程较长,最终会引起锥体束继发性脱髓鞘,经颅磁刺激检查皮层诱发电位引不出,即使出现,CMCT(运动中心传导时间)也明显延长,是正常的2~3倍,而ALS患者CMCT是正常的或轻度延长。这些检查均有助于早期诊断PLS。PLS患者脊髓MRI正常或变细,也有文献报道胸髓MRI T2WI显示皮质脊髓侧束走行区高信号[14],本文有两例患者显示脊髓变细,但未发现脊髓皮质脊髓束走行区异常信号,需继续追踪随访。脑脊液检查正常,也可有蛋白轻度升高。
鉴别诊断:(1)脊髓亚急性联合变性:由于维生素B12缺乏而引起中枢和周围神经系统变性疾病。主要累及脊髓后索、侧索及周围神经,临床表现有双下肢无力、发硬、双手动作笨拙、步态不稳、少数有手套袜套样感觉减退,检查可有双下肢深感觉障碍,Romberg征阳性,常伴贫血。MRI可见脊髓条形、点片状长T1、长T2异常信号。PLS仅有锥体束受累表现,无后索及周围神经受累,可予以鉴别。(2)遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP):该病多在儿童期或青春期发病,临床主要表现为进行性痉挛性截瘫,也可有视神经萎缩、锥体外系症状、共济失调等,儿童期起病可见弓形足,遗传方式主要为常染色体显性遗传、常染色体隐性遗传,少数是X连锁隐性遗传。本文报道的4例患者无家族史,起病较晚,无弓形足,可排除该诊断。(3)多发性硬化:本病多亚急性起病,复发缓解病程,首发症状多为视力下降,MRI髓内有长T1、长T2异常信号,不超过3个椎体节段。本文报道患者均慢性起病,查体发现双侧上运动神经元受损表现,脊髓核磁未见异常,可与该病相鉴别。(4)多系统萎缩(multiple system atrophy,MSA):是一组神经系统变性病,50~60岁多见,临床表现为不同程度的自主神经功能障碍、对左旋多巴反应不良的帕金森综合征、小脑性共济失调和锥体束征等症状。部分患者出现肌萎缩,后期出现肌张力增高,腱反射亢进和病理征。头颅MRI可见壳核背外侧缘条带状弧形高信号,脑桥“十字征”和小脑中脚高信号。直立倾斜试验有体位性低血压,肛门括约肌肌电图出现失神经改变。本文报道患者临床症状与体征目前未见多系统受累的证据,暂不支持该病。
PLS的治疗目前仍然主要是支持治疗,包括康复锻炼,应用药物缓解肌张力、假性球麻痹。PLS预后较ALS好,平均生存时间超过10年[15]。本文4例患者目前生命均未受影响,第1例患者经给予丁苯肽及巴氯芬治疗,蹲起活动较前改善,其余患者病情加重均较缓慢。
综上,对于临床表现为缓慢进展的上运动神经元损害的患者,经详细的病史询问、神经系统查体、实验室及影像学检查排除其他疾病后应高度怀疑PLS,早期诊断,及早治疗,可能会延缓病情发展。未来我们需要更深入的了解PLS发病的病生理机制,为进一步研究该病的防治方法提供指导。
参考文献:
[1] Strickland D, Smith SA, Dolliff G, et al. Amyotrophic lateral sclerosis and occupational history. A pilot case-control study[J]. Arch Neurol, 1996, 53(8): 730-733.
[2] Pringle CE, Hudson AJ, Munoz DG, et al. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria[J]. Brain, 1992, 115 ( Pt 2): 495-520.
[3] Le FN, Maisonobe T, Spelle L, et al. Primary lateral sclerosis: further clarification[J]. J Neurol Sci, 2001, 185(2): 95-100.
[4] Wais V, Rosenbohm A, Petri S, et al. The concept and diagnostic criteria of primary lateral sclerosis[J]. Acta Neurol Scand, 2016, 136(3):204-211.
[5] Gordon PH, Cheng B, Katz IB, et al. The natural history of primary lateral sclerosis[J]. Neurology, 2006, 66:647-653.
[6] Caselli RJ, Smith BE, Osborne D. Primary lateral sclerosis: a neuropsychological study[J]. Neurology, 1995, 45(11): 2005-2009.
[7] Bruyn RP, Koelman JH, Troost D, et al. Motor neuron disease (amyotrophic lateral sclerosis) arising from longstanding primary lateral sclerosis[J]. J Neurol Neurosurg Psychiatry, 1995, 58(6): 742-744.
[8] Mitsumoto H, Nagy PL, Gennings C, et al. Phenotypic and molecular analyses of primary lateral sclerosis[J]. Neurol Genet, 2015, 1(1): e3.
[9] Thorpe JW, Moseley IF, Hawkes CH, et al. Brain and spinal cord MRI in motor neuron disease[J]. J Neurol Neurosurg Psychiatry, 1996, 61(3): 314-317.
[10] Kiernan JA, Hudson AJ. Frontal lobe atrophy in motor neuron diseases[J]. Brain, 1994, 117 ( Pt 4): 747-757.
[11] Agosta F, Canu E, Valsasina P, et al. Divergent brain network connectivity in amyotrophic lateral sclerosis[J]. Neurobiol Aging, 2013, 34(2): 419-427.
[12] Iwata NK, Kwan JY, Danielian LE, et al. White matter alterations differ in primary lateral sclerosis and amyotrophic lateral sclerosis[J]. Brain, 2011, 134(Pt 9): 2642-2655.
[13] Kuipers-Upmeijer J, de Jager AE, Hew JM, et al. Primary lateral sclerosis: clinical, neurophysiological, and magnetic resonance findings[J]. J Neurol Neurosurg Psychiatry, 2001,71(5): 615-620.
[14] Budrewicz S, Szewczyk P, Slotwinski K, et al. Symptoms of degeneration of the pyramidal tracts in conventional magnetic resonance imaging and diffusion tensor imaging in a young woman with primary lateral sclerosis[J]. J Postgrad Med, 2015, 61(3): 206-208.
[15] Floeter MK, Mills R. Progression in primary lateral sclerosis: a prospective analysis[J]. Amyotroph Lateral Scler, 2009, 10(5-6): 339-346.
猜你喜欢
考试与评价·高二版(2020年2期)2020-09-10
实用临床护理学杂志(电子版)(2020年27期)2020-07-15
中西医结合心血管病杂志(电子版)(2020年3期)2020-04-21
实用临床护理学杂志(电子版)(2018年36期)2018-09-13
现代电生理学杂志(2016年3期)2016-07-10
现代电生理学杂志(2016年4期)2016-07-10
中外医疗(2015年16期)2016-01-04
实用手外科杂志(2015年1期)2015-08-27
现代电生理学杂志(2015年3期)2015-07-18
现代电生理学杂志(2015年2期)2015-07-18
