
新型抗真菌药物研究进展
2018-05-16 02:05:02刘昱阎澜姜远英
中国真菌学杂志 2018年2期
刘昱 阎澜 姜远英
(第二军医大学药学院新药研究中心,上海 200433)
侵袭性真菌感染 (invasive fungal infections,IFI)已经成为危害人类健康的严重威胁之一,全球年均死亡超过一百五十万人,而随着免疫抑制剂的使用、肿瘤放化疗、体内置管及久驻ICU病人的增加,IFI逐年上升的发病率与致死率正越来越多地引起人们的关注[1]。药物治疗是应对IFI的主要策略,而有限的药物种类、日渐严重的耐药性和毒副作用等问题,使得研发抗真菌新药的需求变得愈加迫切。
常用的抗真菌药物包括氮唑类、多烯类以及棘白菌素类等[1]。氮唑类是目前最常用的一类抗真菌药物,根据化学结构可分为咪唑类和三氮唑类。咪唑类药物 (咪康唑、酮康唑等)开发最早,抗真菌活性也较高,但由于毒性较大而局限于外用。三氮唑类药物出现较晚,可体内给药治疗IFI。第一代三氮唑类药物 (氟康唑、伊曲康唑等)的抗菌谱较广,但久用易普遍产生耐药性;第二代三氮唑类药物 (伏立康唑、泊沙康唑等)的药效更强,耐药菌株较少见[1-3];最新上市的三氮唑类药物艾沙康唑(isavuconazole)于2015年经FDA批准用于治疗侵袭性曲霉病[4],并已进入治疗侵袭性念珠菌病的III期临床试验。
多烯类药物中,两性霉素B是目前最有效的抗IFI药物[1],但它具有严重的发热、寒战、肾毒性等毒副作用[5]。利用新的两性霉素B剂型,如脂质体[6]、纳米悬浮液[7],以及两性霉素B与阿拉伯半乳聚糖、聚乙二醇等的复合体[8-9]等,可降低两性霉素B的毒副作用。两性霉素B脂质体已在临床上使用,其余剂型尚在研发当中。
临床常用的棘白菌素类药物仅卡泊芬净、米卡芬净、阿尼芬净三种,它们的抗菌谱较窄,对隐球菌无效,且难以通过胃肠道吸收,仅能通过每日1次静脉滴注给药维持体内药物浓度。此外因基因突变导致对棘白菌素耐药真菌的出现,也限制了此类药物的使用[10-11]。
真菌作为真核生物,细胞中可作为药物靶点的基因或蛋白大多与哺乳动物具有同源性,作用于此类靶点的药物易在人体中产生副作用,因此开发具有真菌特异性的抗真菌药物是此类药物研发的关键[12-13]。目前对新型抗真菌药物研发的策略包括:在临床常用药物的基础上改进结构或寻找新结构化合物;寻找新的药物靶点并开发针对新靶点的化合物;从中药成分中寻找有效抗真菌药物或增效剂等。现将部分新型抗真菌药物及其研究进展作一综述。
1 新型氮唑类药物
氮唑类药物的作用机制:通过抑制ERG11基因编码的14α-羊毛甾醇脱甲基酶 (CYP51),抑制真菌细胞膜中的羊毛甾醇转化为麦角甾醇,并使毒性的甾醇产物在真菌细胞中积累,从而抑制真菌的生长与复制[1]。氮唑类药物面临的主要问题包括耐药性的不断扩大、对人体中CYP51的同源酶体 (CYP3A4、CYP2C9等)的抑制所引起的与多种药物相互作用等副反应。为解决上述问题,Viamet Pharmaceuticals公司研发了对真菌具有更高选择性的VT-1161与VT-1129等四氮唑类药物 (见图1a~b)[14]。
1.1 VT-1161
VT-1161对真菌的CYP51具有比其他氮唑类药物更高的选择性,对白念珠菌CYP51的亲和力 (Kd≤39 nmol/L)超过了对人体中同源酶体的2 000倍以上[15]。它能够有效抑制CYP51活性 (半抑制浓度IC50范围1.4~1.6 μmol/L),使白念珠菌中麦角甾醇含量降低95%[15]。VT-1161体外抑制白念珠菌的最低抑菌浓度 (MIC)为0.002 μg/mL,对耐氟康唑菌株的MIC90为0.12 μg/mL[15-16];还可有效作用于小鼠的球孢子菌病 (球孢子菌体外MIC90为2 μg/mL)、皮肤癣菌引起的浅部真菌感染以及根霉菌感染引起的毛霉菌病等感染模型[17-20]。小鼠体内药动学实验显示,VT-1161具有分布体积广 (1.4 L/kg)、口服生物利用度高 (73%)、半衰期长 (>48 h)等特点[16]。VT-1161目前已经完成了治疗 (趾)甲真菌病和复发性阴道念珠菌病的IIb期临床试验[16]。
1.2 VT-1129
VT-1129具有与VT-1161相同的基本骨架,主要表现出对隐球菌,如新生隐球菌 (MIC90为0.060 μg/mL)和格特隐球菌 (MIC90为0.25 μg/mL)等的抑制活性[21],对耐氟康唑的新生隐球菌具有一定的抑制作用 (MIC50为0.25 μg/mL ,MIC90为2 μg/mL)[22]。VT-1129对隐球菌的CYP51具有强亲和力及选择性抑制作用 (Kd值为14~25 nmol/L,IC50范围0.14~0.20 μmol/L),对人体中CYP51及同源酶体的亲和力和抑制作用较弱 (Kd为4.53 μmol/L,IC50约为600 μmol/L)[23]。Alexander等人的研究还证实了VT-1161与VT-1129对光滑念珠菌以及克柔念珠菌的耐药菌株具有抑制活性[24]。目前已开展了VT-1129用于治疗隐球菌性脑膜炎的I期临床试验。
此外,Viamet Pharmaceuticals公司最新研发的又一具有广谱抗真菌作用的四氮唑类化合物VT-1598 (见图1c),显示出对酵母菌、丝状真菌等具有体外抑制活性,还能抑制耳念珠菌等耐药真菌的生长[25-26],即将进入针对包括隐球菌性脑膜炎在内的一系列侵袭性真菌感染的临床试验。
2 新型葡聚糖合成酶抑制剂
β-1,3-葡聚糖是真菌细胞壁的重要组成成分,构成了50%以上的细胞壁,并连接壳聚糖和糖蛋白等其他细胞壁组分,抑制其合成有助于破坏真菌细胞壁,促进真菌细胞凋亡。棘白菌素类是最常用的β-1,3-葡聚糖合成酶的抑制剂,但半衰期短、口服生物利用度低等缺点 (例如常用的棘白菌素类药物中最稳定的阿尼芬净的半衰期仅30 h),加上耐棘白菌素菌株的出现,共同限制了它们的临床使用[11]。目前研发的新型β-1,3-葡聚糖合成酶抑制剂中,具有代表性的包括长效棘白菌素类药物CD101与非棘白菌素类药物SCY-078等。
2.1 CD101
CD101 (Cidara Therapeutics Inc.,见图2a)是目前最长效的一种棘白菌素类药物,在人体内的终末半衰期可达到约133 h[27-28]。CD101的体内外抗真菌活性与其他棘白菌素类药物相似[29-35],能够有效抑制念珠菌 (白念珠菌和热带念珠菌的MIC90为0.06 μg/mL)和曲霉菌 (烟曲霉和黄曲霉的最低有效浓度MEC90为0.03 μg/mL),但对隐球菌无效[31-33]。CD101在棘白菌素类药物中具有更好的稳定性,不加入稳定剂的条件下,在37℃的人类血浆中培养44 h后,可检测到的残余量为93%,而阿尼芬净可检测到的残余量仅为7%[36]。棘白菌素类药物体内代谢产生的中间体可能是造成肝毒性的主要因素,CD101的高稳定性使其在人体内的代谢分解及中间体的生成减少,因而几乎不产生肝毒性[34]。I期临床试验显示,CD101在健康成年人体内具有低累积量 (30%~55%)、低表观清除率 (<0.28 L/h)和半衰期长 (>80 h)、组织分布广等特点[28]。此外,给药过程的差异能够影响CD101的抗真菌效果,总剂量相同时,每周1次给药比每周2次或每日1次表现出更好的杀菌作用[37]。CD101静脉注射剂 (rezafungin)目前已经完成II期临床试验,用于治疗念珠菌血症并局部用于治疗初发或复发性阴道念珠菌病[31]。
2.2 SCY-078
SCY-078 (原名MK-3118,Scynexis Inc.,见图2b)是天然化合物Enfumafungin[38-39](提取自植物内生真菌Hormonemasp.)的半合成衍生物,具有不同于棘白菌素的三萜烯结构[38],是一种新型的口服β-1,3-葡聚糖合成酶抑制剂[40]。SCY-078在体内外均能发挥对念珠菌的抑制作用[41-42],对念珠菌或曲霉菌的抑制活性略低于棘白菌素[38,42-44],但对耐氮唑类药物菌株及fsk基因突变引起的耐棘白菌素菌株活性较强[43,45-46],对非曲霉菌属丝状真菌的活性多与棘白菌素相似,对多育赛多孢的活性略强于其他药物[47],此外还证实了SCY-078对耳念珠菌具有抗生物被膜形成的活性 (MIC90为1 μg/mL)[48]。药动学研究[49]表明,SCY-078具有良好的口服生物利用度,体外利用Caco-2单层细胞评价其口服肠吸收的渗透性,5 μmol/L的SCY-078从肠腔侧到基底侧方向的表观渗透速率为 (8.9±0.78)×10-6cm/s (>10-6cm/s即可认为口服吸收性良好)。SCY-078在小鼠、大鼠和犬类中的口服生物利用度分别为>51%、45%和35%,口服半衰期分别为8.3、9.1和15.2 h,肾组织暴露量是血浆暴露量的20~25倍,肾分布率高于棘白菌素类[49]。SCY-078目前已经进入治疗侵袭性真菌病的II期临床试验[49]。
由于棘白菌素类药物只能通过静脉滴注给药,因此能够口服给药的SCY-078未来在临床使用上将具有独特的优势。
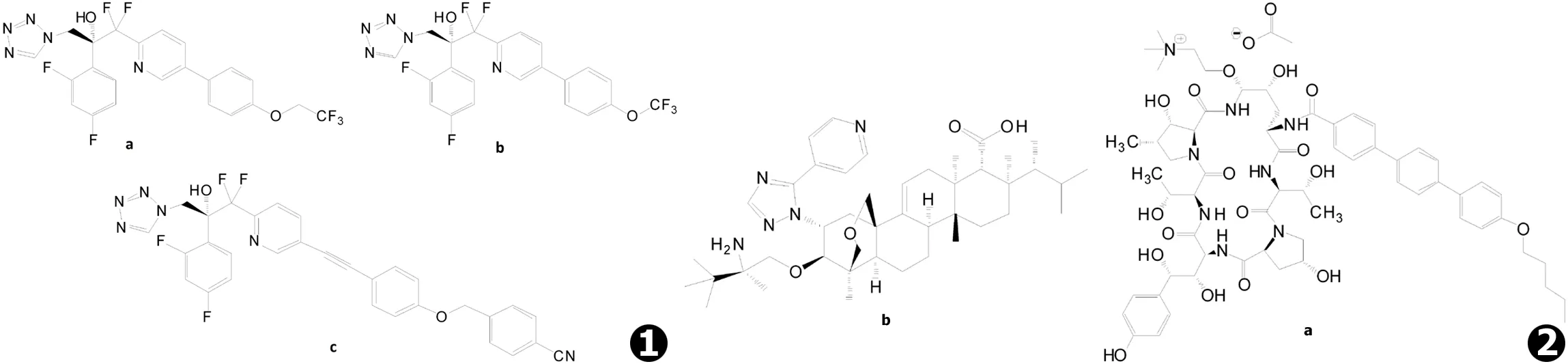
图1 新型氮唑类药物:a.VT-1161,b.VT-1129,c.VT-1598 图2 新型葡聚糖合成酶抑制剂:a.CD101,b.SCY-078 (MK-3118)
3 新靶点抗真菌药物
3.1 GPI锚定蛋白抑制剂E1210
细胞表面的蛋白在真菌的生命活动中发挥着重要作用,其中的部分功能蛋白属于糖基磷脂酰肌醇 (glycosyl phosphatidyl inositol,GPI)锚定蛋白,即被GPI锚定于细胞膜外表面或交叉结合于真菌细胞壁的甘露糖蛋白[50]。尽管尚有超过半数的真菌GPI锚定蛋白的功能未知,但目前已知部分GPI锚定蛋白对真菌细胞壁的再生及对宿主细胞的黏附发挥着必不可少的作用[50],因此有望将GPI锚定蛋白的生物合成途径作为治疗真菌感染的新靶点[51-52]。
GPI锚定蛋白抑制剂E1210 (日本卫材制药,又名APX001A,见图3a)能够选择性抑制真菌的GWT1基因编码的肌醇酰基转移酶Gwt1p蛋白,从而抑制GPI合成通路上游的GlcN-PI肌醇酰化反应,但对人类GWT1同源基因PIG-W编码的蛋白无抑制作用[51-53]。E1210通过抑制GlcN-PI的肌醇酰化反应抑制GPI的合成,导致GPI锚定蛋白的成熟和转运过程的缺陷,使得细胞表面GPI锚定蛋白含量水平降低,从而抑制了真菌的黏附作用、菌丝生长和生物被膜形成等与真菌毒力相关的生命过程[53]。体外药敏实验[54-58]显示E1210对念珠菌 (MIC90为0.06 μg/mL)[55]、曲霉菌 (MEC90≤0.06 μg/mL)[56]、镰刀菌以及丝孢菌 (MEC90为0.12 μg/mL)[57]等具有显著的抑制作用,并能够抑制耐氮唑类或棘白菌素类药物的白念珠菌和曲霉菌[54-56,58]。体内活性方面,口服E1210能够显著减少口咽念珠菌病小鼠口腔中念珠菌的数量,提高播散性念珠菌病、肺曲霉病以及播散性珠镰刀菌病小鼠的生存率,上述作用具有剂量依赖性,发挥药效需要较高的体内浓度[59]。相比于令人瞩目的体外抗真菌结果,E1210的体内利用则稍弱于体外作用。药动学实验显示,E1210静脉给药后的血浆消除半衰期为2.2 h,与伏立康唑近似,但比其他阳性药物短约2~5倍;口服给药0.5 h后可达到最大吸收浓度,口服生物利用度为57.5%[59],相比其他口服药物略显不足,这使得E1210的临床应用将受到一定的限制。目前,E1210的前药APX001已由Amplyx Pharmaceuticals公司完成了I期临床试验,将启动针对侵袭性曲霉病与侵袭性念珠菌病的II期临床项目。
3.2 作用于嘧啶生物合成通路的F901318
细胞中的嘧啶不仅用于合成DNA与RNA,而且能够用作脂质和碳水化合物合成的前体物质,是细胞构成中的必要成分。有研究表明,阻断真菌中嘧啶的生物合成通路能够减弱真菌的毒力,可作为真菌治疗的新策略[60]。
二氢乳清酸脱氢酶 (DHODH)能够催化嘧啶生物合成的第4步反应,即二氢乳清酸转化为乳清酸的反应,是嘧啶生物合成通路中的关键酶[60]。F901318 (F2G Ltd.,见图3b)是真菌DHODH的选择性抑制剂,通过特异性抑制真菌的DHODH (IC50为44±10 nmol/L (n=11))阻断真菌细胞中嘧啶的生物合成通路,发挥抗真菌作用[60]。尽管哺乳动物体内也存在DHODH,但由于与真菌的同源性较低,F901318在体外对烟曲霉DHODH的作用超过对人体DHODH的作用2 200倍以上 (IC50>100 μmol/L)[60]。酶动力学实验显示,F901318是烟曲霉DHODH的可逆性抑制剂,同时也是与辅酶Q辅因子有关的竞争性抑制剂[60]。F901318对烟曲霉 (MIC90为0.125 μg/mL)等曲霉菌以及青霉菌、粗球孢子菌、荚膜组织胞浆菌、皮炎芽生菌、镰刀菌以及多育赛多孢等多种致病性丝状真菌或二态真菌具有抑制作用,但对念珠菌和接合菌等未检测到体外活性,可能由F901318独特的靶点所致[60-61]。由于F901318的作用靶点不同于氮唑类药物,因此其作用不受Cyp51A基因突变所引起的氮唑类耐药的影响[60]。目前F901318已完成了口服与静脉注射治疗侵袭性曲霉病的I期临床试验,进入II期临床试验阶段。
4 抗真菌中药及增效剂
中药是我国传统文化的瑰宝,从中药中寻找具有抗真菌活性的有效成分是新药发现的重要方式之一。抗真菌增效剂的开发已经成为抗真菌新药开发的又一常用手段,中药成分除直接作用于真菌外,也可作为增效剂,与其他药物联合用药发挥协同抗真菌作用。本课题组通过对中药有效成分的筛选,发现了能够影响白念珠菌线粒体氧化呼吸的紫草素[62]、能够抗白念珠菌生物被膜形成的紫檀茋[63-64]和汉防己甲素[65]等具有抗真菌作用的中药成分,以及小檗碱[66-69]、黄芩素[70-72]、光甘草定[73]、蛇床子素[74]、覆盆子提取物[75]和月季花提取物[76]等具有抗真菌增效剂作用的中药成分。
本课题组对小檗碱协同氟康唑等发挥抗真菌作用的机制开展了较为系统的研究。小檗碱 (见图4a)又名黄连素,是中药黄连中的主要生物碱,它单独作用于耐药白念珠菌的活性较弱 (MIC80>32 μg/mL),与氟康唑合用则能够发挥协同抗真菌作用,合用后氟康唑与小檗碱抑制耐药白念珠菌的平均MIC80分别降低至1 μg/mL与2 μg/mL,分数抑制浓度指数 (FICI)范围0.017~0.127[66]。小檗碱与氟康唑合用能够引起耐药白念珠菌核DNA损伤并影响其细胞周期,时间-杀菌曲线显示二者的协同作用表现出对小檗碱的剂量依赖性,说明二者合用时小檗碱可能发挥了主要作用,已有研究证实小檗碱能够结合于DNA并影响DNA的复制与转录并破坏细胞周期,荧光实验显示小檗碱可在白念珠菌细胞内尤其是细胞核内累积,而氟康唑可破坏真菌细胞膜的完整性,因此猜想细胞膜的破坏能够增加小檗碱进入细胞中的浓度,从而增强其对耐药真菌的作用[68]。除氟康唑外,小檗碱与酮康唑、伊曲康唑及两性霉素B等作用于细胞膜的药物均可发挥协同作用,与5-氟尿嘧啶、卡泊芬净等则无协同作用,进一步支持了上述猜想[68]。后续研究还发现,小檗碱可通过结合至相应的药物反应元件 (DRE),阻断白念珠菌耐药基因CDR1的转录启动过程,从而增强氟康唑抗耐药白念珠菌的作用[69],这一发现提示氟康唑与小檗碱能够相互促进对方的抗真菌作用。此外,比较蛋白质组学研究显示,氟康唑合用小檗碱可调节线粒体有氧呼吸并引起内源性活性氧 (ROS)的增加,可能有助于协同作用的发挥[67]。相关机制有待更为深入的研究。
黄芩素 (见图4b)是中药黄芩的有效成分,具有多种药理活性。黄芩素单独作用于耐药白念珠菌的作用较弱 (MIC80>64 μg/mL),与氟康唑合用后可使氟康唑对耐药白念珠菌的MIC80平均降低256倍,同时自身对耐药白念珠菌的平均MIC80降至2 μg/mL,FICI范围为0.037~0.098[70]。黄芩素与氟康唑合用对白念珠菌生物被膜具有抑制作用,二者合用影响白念珠菌与细胞表面疏水性、菌丝形成及黏附作用相关基因的表达,使得细胞表面疏水性降低、菌丝形成减少,从而降低细胞黏附能力[71]。此外还发现黄芩素单独使用具有破坏白念珠菌的线粒体膜电位、诱导细胞凋亡的作用[72]。
5 其他抗真菌新药
T-2307 (日本富山化工,见图5a)是新型的芳基嘧啶类抗真菌化合物,在体外对包括耐氟康唑菌株在内的念珠菌 (MIC范围0.000 25~0.007 8 μg/mL)、隐球菌 (MIC范围0.003 9~0.062 5 μg/mL)与曲霉菌 (MIC范围0.015 6~4 μg/mL)等均表现出良好的抗真菌活性,体内活性近似或优于其他常用抗真菌药物[77-80]。T-2307注射剂已进行了I期临床试验。研究表明,T-2307可能通过作用于真菌的线粒体、破坏氧化呼吸链发挥对真菌的抑制作用,而T-2307对真菌线粒体的亲和力是对哺乳动物的500倍,若这一机制成立,则T-2307有望成为具有选择性的新靶点抗真菌药物[81-82]。
尼可霉素Z (Valley Fever Solutions, Inc.,见图5b)是一种几丁质合成酶抑制剂,通过干扰真菌细胞壁中几丁质的合成抑制真菌生长[83-84],目前将进入治疗球孢子菌病的II期临床试验。但同类药物的水解倾向以及体内活性不足等缺点将是限制尼可霉素Z临床应用的主要因素[1]。
VL-2397 (又名ASP2397,Astellas Pharma Inc.,见图5c)是利用线虫感染模型从枝顶孢属真菌的天然产物中筛选得到的抗真菌活性化合物,对曲霉菌具有较强抑制活性而对念珠菌无活性,对野生型和Cyp51A基因突变的耐氮唑类药物烟曲霉的MIC50分别为0.25 μg/mL和0.125 μg/mL,其抗真菌药效与两性霉素B接近,但作用机制尚未明确[85-87],目前已进入治疗侵袭性曲霉病的II期临床试验阶段。
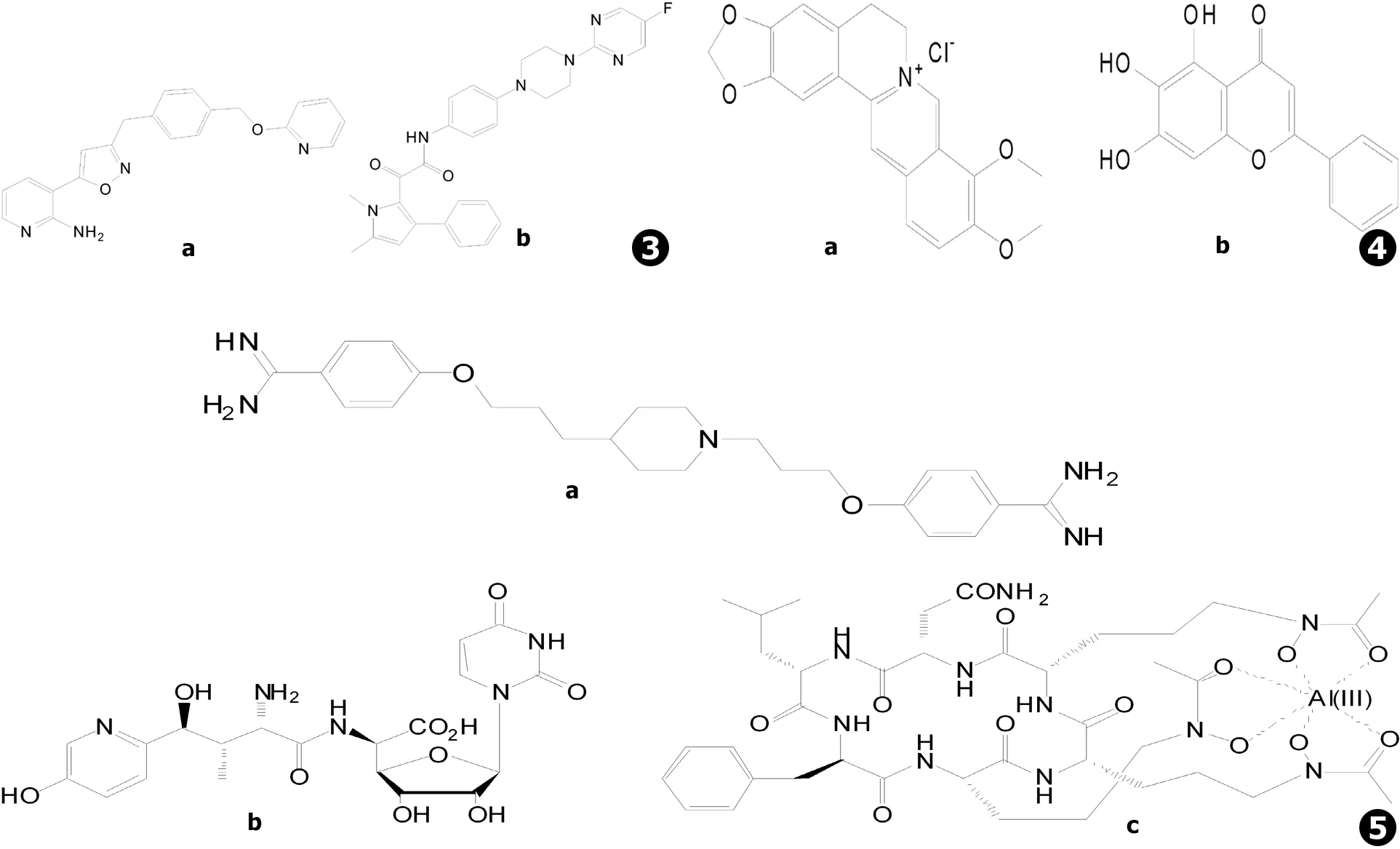
图3新靶点抗真菌药物:a.E1210 (AX001),b.F901318图4抗真菌中药及增效剂:a.盐酸小檗碱,b.黄芩素图5其他抗真菌新药:a.T-2307,b.尼可霉素Z,c.VL-2397 (ASP2397)
Fig.3New target antifungal agents:a.E1210 (AX001),b.F901318Fig.4Traditional Chinese medicine antifungal synergists:a.Berberine hydrochloride,b.BaicaleinFig.5Other novel antifungal agents:a.T-2307,b.Nikkomycin Z,c.VL-2397 (ASP2397)
6 总结与展望
经过长期的临床实践的检验,氮唑类、棘白菌素类等广泛使用的抗真菌药物的有效性与安全性已经得到了验证,作用机制的研究也比较深入。在此基础上通过改进结构等方法产生更安全有效的抗真菌新药,仍是抗真菌新药研发的重要方式。对真菌特异性新靶点的发现,正在为抗真菌药物的研发提供新的思路,据此已经发现了数个新的抗真菌药物靶点,并产生了基于新靶点的抗真菌药物。此外,从中药中寻找有效抗真菌药物或增效剂正成为新药研发的又一重要手段。随着研究的深入,未来将涌现出更多值得人们关注的新型抗真菌药物,为临床治疗侵袭性真菌感染提供更多可供选择的药物治疗方案。
[1] Campoy S,Adrio JL.Antifungals[J].Biochem Pharmacol,2017,133: 86-96.
[2] Peyton LR,Gallagher S,Hashemzadeh M.Triazole antifungals: a review[J].Drugs Today (Barc),2015,51(12):705-718.
[3] Moore JN,Healy JR,Kraft WK.Pharmacologic and clinical evaluation of posaconazole[J].Expert Rev Clin Pharmacol,2015,83(3):321-334.
[4] Shirley M,Scott LJ.Isavuconazole: a review in invasive aspergillosis and mucormycosis[J].Drugs,2016,76(17):1647-1657.
[5] Osherov N,Kontoyiannis DP.The anti-Aspergillusdrug pipeline: is the glass half full or empty?[J].Med Mycol,2017,55(1):118-124.
[6] Torrado JJ,Espada R,Ballesteros MP,et al.Amphotericin B formulations and drug targeting[J].J Pharm Sci, 2008,97(7): 2405-2425.
[7] Halperin A,Shadkchan Y,Pisarevsky E,et al.Novel watersoluble amphotericin B-PEG conjugates with low toxicity and potentinvivoefficacy[J]. J Med Chem,2016,59(3):1197-1206.
[8] Ickowicz DE,Farber S,Sionov E,et al.Activity, reduced toxicity, and scale-up synthesis of amphotericin B-conjugated polysaccharide[J]. Biomacromolecules,2014,15(6):2079-2089.
[9] Shirkhani K,Teo I,Armstrong-James D,et al.Nebulised amphotericin B-polymethacrylic acid nanoparticle prophylaxis prevents invasive aspergillosis[J].Nanomedicine,2015,11(5):1217-1226.
[10] Denning DW.Echinocandin antifungal drugs[J].Lancet,2003,362(9390):1142-1151.
[11] Chang CC,Slavin MA,Chen SC.New developments and directions in the clinical application of the echinocandins[J].Arch Toxicol,2017,91(4):1613-1621.
[12] Roemer T,Krysan DJ.Antifungal drug development: challenges, unmet clinical needs, and new approaches[J]. Cold Spring Harb Perspect Med,2014,4(5):a019703.
[13] Denning DW,BromLey MJ.Infectious Disease. How to bolster the antifungal pipeline[J].Science,2015,347(6229):1414-1416.
[14] Hoekstra WJ,Garvey EP,Moore WR,et al.Design and optimization of highly-selective fungal CYP51 inhibitors[J].Bioorg Med Chem Lett,2014,24(15):3455-3458.
[15] Warrilow AGS,Martel CM,Parker JE,et al.The clinical candidate VT-1161 is a highly potent inhibitor ofCandidaalbicansCYP51 but fails to bind the human enzyme[J].Antimicrob Agents Chemother,2014,58(12):7121-7127.
[16] Garvey EP,Hoekstra WJ,Schotzinger RJ,et al.Efficacy of the clinical agent VT-1161 against fluconazole-sensitive and -resistantCandidaalbicansin a murine model of vaginal candidiasis[J].Antimicrob Agents Chemother,2015,59(9):5567-5573.
[17] Shubitz LF,Trinh HT,Galgiani JN,et al.Evaluation of VT-1161 for treatment of coccidioidomycosis in murine infection models[J].Antimicrob Agents Chemother,2015,59(12):7249-7254.
[18] Shubitz LF,Roy ME,Trinh HT,et al.Efficacy of the investigational antifungal VT-1161 in treating naturally occurring Coccidioidomycosis in dogs[J].Antimicrob Agents Chemother,2017,61(5):e00111-17.
[19] Garvey EP,Hoekstra WJ,Moore WR,et al.VT-1161 dosed once daily or once weekly exhibits potent efficacy in treatment of dermatophytosis in a guinea pig model[J].Antimicrob Agents Chemother,2015,59(4):1992-1997.
[20] Gebremariam T,Wiederhold NP,Fothergill AW,et al.VT-1161 protects immunosuppressed mice fromRhizopusarrhizusvar.arrhizusinfection[J].Antimicrob Agents Chemother,2015,59(12):7815-7817.
[21] Lockhart SR,Fothergill AW,Wiederhold NP,et al. The investigational fungal Cyp51 inhibitor VT-1129 demonstrates potentinvitroactivity againstCryptococcusneoformansandCryptococcusgattii[J].Antimicrob Agents Chemother,2016,60(4):2528-2531.
[22] Nielsen K,Vedula P,Smith KD,et al.Activity of VT-1129 againstCryptococcusneoformansclinical isolates with high fluconazole MICs[J].Med Mycol,2017,55(4):453-456.
[23] Warrilow AGS,Parker JE,Price CL,et al.The investigational drug VT-1129 is a highly potent inhibitor ofCryptococcusspecies CYP51 but only weakly inhibits the human enzyme[J].Antimicrob Agents Chemother,2016,60(8):4530-4538.
[24] Schell WA,Jones AM,Garvey EP,et al.Fungal CYP51 Inhibitors VT-1161 and VT-1129 exhibit stronginvitroactivity againstCandidaglabrataandC.kruseiisolates clinically resistant to azole and echinocandin antifungal compounds[J].Antimicrob Agents Chemother,2017,61(3):e01817-16.
[25] Hargrove TY,Garvey EP,Hoekstra WJ,et al. Crystal structure of the new investigational drug candidate VT-1598 in complex withAspergillusfumigatussterol 14α-Demethylase provides insights into its broad-spectrum antifungal activity[J].Antimicrob Agents Chemother,2017,61(7):e00570-17.
[26] Wiederhold NP,Patterson HP,Tran BH,et al. Fungal-specific Cyp51 inhibitor VT-1598 demonstratesinvitroactivity against Candida and Cryptococcus species, endemic fungi, including Coccidioides species, Aspergillus species and Rhizopus arrhizus[J]. J Antimicrob Chemother,2018,73(2):404-408.
[27] Ong V,James KD,Smith S,et al.Pharmacokinetics of the novel echinocandin CD101 in multiple animal species[J].Antimicrob Agents Chemother,2017,61(4):e01626-16.
[28] Sandison T,Ong V,Lee J,et al.Safety and pharmacokinetics of CD101 IV, a novel echinocandin, in healthy adults[J].Antimicrob Agents Chemother,2017,61(2):e01627-16.
[29] Zhao Y, Perez WB,Jiménez-Ortigosa C,et al. CD101: a novel long-acting echinocandin[J].Cell Microbiol,2016,18(9):1308-1316.
[30] Pfaller MA,Messer SA,Rhomberg PR,et al.Activity of a long-acting echinocandin, CD101, determined using CLSI and EUCAST reference methods, against Candida and Aspergillus spp., including echinocandin- and azole-resistant isolates[J].J Antimicrob Chemother,2016,71(10):2868-2873.
[31] James KD,Laudeman CP,Malkar NB,et al. Structure-Activity Relationships of a Series of Echinocandins and the Discovery of CD101, a Highly Stable and Soluble Echinocandin with Distinctive Pharmacokinetic Properties[J].Antimicrob Agents Chemother,2017,61(2):e01541-16.
[32] Pfaller MA,Messer SA,Rhomberg PR,et al. Activity of a long-acting echinocandin (CD101) and seven comparator antifungal agents tested against a global collection of contemporary invasive fungal isolates in the SENTRY 2014 Antifungal Surveillance Program[J].Antimicrob Agents Chemother,2017,61(3):e02045-16.
[33] Pfaller MA,Messer SA,Rhomberg PR,et al.CD101, a long-acting echinocandin, and comparator antifungal agents tested against a global collection of invasive fungal isolates in the SENTRY 2015 Antifungal Surveillance Program[J].Int J Antimicrob Agents,2017,50(3):352-358.
[34] Ong V,Hough G,Schlosser M,et al.Preclinical evaluation of the stability, safety, and efficacy of CD101, a novel echinocandin[J].Antimicrob Agents Chemother,2016,60(11):6872-6879.
[35] Zhao Y,Prideaux B, Nagasaki Y,et al. Unraveling Drug Penetration of Echinocandin Antifungals at the Site of Infection in an Intra-abdominal Abscess Model[J].Antimicrob Agents Chemother,2017,61(10):e01009-17.
[36] Krishnan BR,James KD,Polowy K,et al.CD101, a novel echinocandin with exceptional stability properties and enhanced aqueous solubility[J].J Antibiot (Tokyo),2017,70(2):130-135.
[37] Lakota EA,Bader JC,Ong V,et al.Pharmacological basis of CD101 efficacy: exposure shape matters[J]. Antimicrob Agents Chemother,2017,61(11):e00758-17.
[38] Hector RF,Bierer DE.New β-glucan inhibitors as antifungal drugs[J]. Expert Opin Ther Pat,2011,21(10):1597-1610.
[39] Onishi J,Meinz M,Thompson J,et al.Discovery of novel antifungal (1,3)-beta-D-glucan synthase inhibitors[J]. Antimicrob Agents Chemother,2000,44(2):368-377.
[40] Scorneaux B,Angulo D,Borroto-Esoda K,et al.SCY-078 is fungicidal against Candida species in time-kill studies[J].Antimicrob Agents Chemother,2017,61(3):e01961-16.
[41] Lepak AJ,Marchillo K,Andes DR.Pharmacodynamic target evaluation of a novel oral glucan synthase inhibitor, SCY-078 (MK-3118), using aninvivomurine invasive candidiasis model[J].Antimicrob Agents Chemother,2015,59(2):1265-1272.
[42] Schell WA,Jones AM,Borroto-Esoda K,et al.Antifungal activity of SCY-078 and standard antifungal agents against 178 clinical isolates of resistant and susceptibleCandidaspecies[J].Antimicrob Agents Chemother,2017,61(11):e01102-17.
[43] Pfaller MA,Messer SA,Motyl MR,et al.Activity of MK-3118, a new oral glucan synthase inhibitor, tested againstCandidaspp. by two international methods (CLSI and EUCAST)[J]. J Antimicrob Chemother,2013,68(4):858-863.
[44] Pfaller MA,Messer SA,Motyl MR,et al.Invitroactivitity of a new oral glucan synthase inhibitor (MK-3118) tested againstAspergillusspp. by CLSI and EUCAST broth microdilution methods[J].Antimicrob. Agents Chemother,2013,57(2):1065-1068.
[45] Jimenez-Ortigosa C,Paderu P,Motyl MR,et al.Enfumafungin derivative MK-3118 shows increasedinvitropotency against clinical echinocandin-resistantCandidaspecies andAspergillusspecies isolates[J].Antimicrob Agents Chemother,2014,58(2):1248-1251.
[46] Pfaller MA,Messer SA,Rhomberg PR,et al.Differential activity of the oral glucan synthase inhibitor SCY-078 against wild-type and echinocandin-resistant strains ofCandidaspecies[J].Antimicrob Agents Chemother,2017,61(8):e00161-17.
[47] Lamoth F,Alexander BD.Antifungal activities of SCY-078 (MK-3118) and standard antifungal agents against clinical non-Aspergillus mold isolates[J].Antimicrob Agents Chemother,2015,59(7):4308-4311.
[48] Larkin E,Hager C,Chandra J,et al.The emerging pathogen Candida auris: growth phenotype, virulence factors, activity of antifungals, and effect of SCY-078, a novel glucan synthesis inhibitor, on growth morphology and biofilm formation[J].Antimicrob Agents Chemother,2017,61(5):e02396-16.
[49] Wring SA,Randolph R,Park S,et al.Preclinical pharmacokinetics and pharmacodynamic target of SCY-078, a first-in-class orally active antifungal glucan synthesis inhibitor, in murine models of disseminated candidiasis[J].Antimicrob Agents Chemother,2017,61(4):e02068-16.
[50] Richard ML,Plaine A.Comprehensive analysis of glycosylphosphatidylinositol-anchored proteins inCandidaalbicans[J].Eukaryot Cell,2007,6(2):119-133.
[51] Tsukahara K,Hata K,Nakamoto K,et al.Medicinal genetics approach towards identifying the molecular target of a novel inhibitor of fungal cell wall assembly[J].Mol Microbiol,2003,48(4):1029-1042.
[52] Umemura M,Okamoto M,Nakayama K,et al.GWT1 gene is required for inositol acylation of glycosylphosphatidylinositol anchors in yeast[J]. J Biol Chem,2003,278(26):23639-23647.
[53] Watanabe NA,Miyazaki M,Horii T,et al.E1210, a new broad-spectrum antifungal, suppressesCandidaalbicanshyphal growth through inhibition of glycosylphosphatidylinositol biosynthesis[J].Antimicrob. Agents Chemother,2012,56(2):960-971.
[54] Miyazaki M,Horii T,Hata K,et al.Invitroactivity of E1210, a novel antifungal, against clinically important yeasts and molds[J].Antimicrob Agents Chemother,2011,55(10): 4652-4658.
[55] Pfaller MA,Hata K,Jones RN,et al.Invitroactivity of a novel broad-spectrum antifungal, E1210, tested againstCandidaspp. as determined by CLSI broth microdilution method[J].Diagn Microbiol Infect Dis,2011,71(2):167-170.
[56] Pfaller MA,Duncanson F,Messer SA,et al.Invitroactivity of a novel broad-spectrum antifungal, E1210, tested againstAspergillusspp. determined by CLSI and EUCAST broth microdilution methods[J].Antimicrob Agents Chemother,2011,55(11):5155-5158.
[57] Castanheira M,Duncanson FP,Diekema DJ,et al.Activities of E1210 and comparator agents tested by CLSI and EUCAST broth microdilution methods againstFusariumandScedosporiumspecies identified using molecular methods[J].Antimicrob Agents Chemother,2012,56(1):352-357.
[58] Wiederhold NP,Najvar LK,Fothergill AW,et al.The investigational agent E1210 is effective in treatment of experimental invasive candidiasis caused by resistantCandidaalbicans[J].Antimicrob Agents Chemother,2015,59(1):690-692.
[59] Hata K,Horii T,Miyazaki M,et al.Efficacy of oral E1210, a new broad-spectrum antifungal with a novel mechanism of action, in murine models of candidiasis, aspergillosis, and fusariosis[J].Antimicrob Agents Chemother,2011,55(10):4543-4551.
[60] Oliver JD,Sibley GE,Beckmann N,et al.F901318 represents a novel class of antifungal drug that inhibits dihydroorotate dehydrogenase[J].Proc Natl Acad Sci U S A,2016,113(45):12809-12814.
[61] Buil JB,Rijs AJMM,Meis JF,et al.Invitroactivity of the novel antifungal compound F901318 against difficult-to-treatAspergillusisolates[J].J Antimicrob Chemother,2017,72(9):2548-2552.
[62] Miao H,Jiang Y,Cao Y,et al.Inhibitory effect of Shikonin onCandidaalbicansgrowth[J].Biol Pharm Bull, 2012,35(11):1956-1963.
[63] Li DD,Zhao LX,Jiang YY,et al.Invitroandinvivoactivities of pterostilbene againstCandidaalbicansbiofilms[J].Antimicrob Agents Chemother,2014,58(4):2344-2355.
[64] Hu DD,Zhang RL,Jiang YY,et al.The structure-activity relationship of pterostilbene againstCandidaalbicansbiofilms[J]. Molecules, 2017,22(3):E360.
[65] Zhao LX,Li DD,Jiang YY et al.Effect of tetrandrine againstCandidaalbicansbiofilms[J].PLoS One,2013,8(11):e79671.
[66] Quan H,Cao YY,Jiang YY,et al.Potentinvitrosynergism of fluconazole and berberine chloride against clinical isolates ofCandidaalbicansresistant to fluconazole[J].Antimicrob Agents Chemother,2006,50(3):1096-1099.
[67] Xu Y,Wang Y,Jiang YY,et al.Proteomic analysis reveals a synergistic mechanism of fluconazole and berberine against fluconazole-resistantCandidaalbicans: endogenous ROS augmentation[J]. J Proteome Res,2009,8(11):5296-5304.
[68] Li DD,Xu Y,Jiang YY,et al.Fluconazole assists berberine to kill fluconazole-resistantCandidaalbicans[J]. Antimicrob Agents Chemother,2013,57(12):6016-6027.
[69] Zhu SL,Yan L,Jiang YY,et al.Berberine inhibits fluphenazine-induced up-regulation of CDR1 inCandidaalbicans[J].Biol Pharm Bull,2014, 37(2):268-273.
[70] Huang S,Cao YY,Jiang YY,et al.Invitrosynergism of fluconazole and baicalein against clinical isolates ofCandidaalbicansresistant to fluconazole[J].Biol Pharm Bull,2008,31(12):2234-2236.
[71] 赵柳娅,蒋京辰,姚响文,等.黄芩素与氟康唑协同抗白念珠菌生物被膜作用研究[J].中国真菌学杂志,2014,9(2):70-74.
[72] Dai BD,Cao YY,Jiang YY,et al.Baicalein induces programmed cell death inCandidaalbicans[J].J Microbiol Biotechnol,2009,19(8):803-809.
[73] Liu W,Li LP,Jiang YY,et al.Synergistic antifungal effect of glabridin and fluconazole[J].PLoS One,2014,9(7):e103442.
[74] Li DD,Chai D,Jiang YY.Potentinvitrosynergism of fluconazole and osthole against fluconazole-resistantCandidaalbicans[J].Antimicrob Agents Chemother,2017,61(8):e00436-17.
[75] Han B,Cao YB,Jiang YY,et al.Antifungal activity ofRubuschingiiextract combined with fluconazole against fluconazole-resistantCandidaalbicans[J].Microbiol Immunol,2016,60(2):82-92.
[76] Zhang L,Lin H,Jiang YY,et al.Antifungal activity of the ethanol extract from Flos Rosae Chinensis with activity against fluconazole-resistant clinicalCandida[J].Evid Based Complement Alternat Med,2017,2017:4780746.
[77] Mitsuyama J,Nomura N,Hashimoto K,et al.Invitroandinvivoantifungal activities of T-2307, a novel arylamidine[J].Antimicrob Agents Chemother,2008,52(4):1318-1324.
[78] Wiederhold NP,Najvar LK,Fothergill AW,et al.The novel arylamidine T-2307 maintainsinvitroandinvivoactivity against echinocandin-resistantCandidaalbicans[J].Antimicrob Agents Chemother,2015,59(2):1341-1343.
[79] Wiederhold NP,Najvar LK,Fothergill AW,et al.The novel arylamidine T-2307 demonstratesinvitroandinvivoactivity against echinocandin-resistantCandidaglabrata[J].J Antimicrob Chemother,2016,71(3): 692-695.
[80] Nishikawa H,Fukuda Y,Mitsuyama J,et al.Invitroandinvivoantifungal activities of T-2307, a novel arylamidine, againstCryptococcusgattii: an emerging fungal pathogen[J]. J Antimicrob Chemother,2017,72(6):1709-1713.
[81] Yamada E,Nishikawa H,Nomura N,et al.T-2307 shows efficacy in murine model ofCandidaglabratainfection despiteinvitrotrailing growth phenomena[J].Antimicrob Agents Chemother,2010,54(9):3630-3634.
[82] Shibata T,Takahashi T,Yamada E,et al.T-2307 causes collapse of mitochondrial membrane potential in yeast[J].Antimicrob Agents Chemother,2012,56(11):5892-5897.
[83] Holden WM,Fites JS,Reinert LK,et al.Nikkomycin Z is an effective inhibitor of the chytrid fungus linked to global amphibian declines[J].Fungal Biol,2014,118(1):48-60.
[84] Shubitz LF,Trinh HT,Perrill RH,et al.Modeling nikkomycin Z dosing and pharmacology in murine pulmonary coccidioidomycosis preparatory to phase 2 clinical trials[J].J Infect Dis,2014,209(12):1949-1954.
[85] Arendrup MC,Jensen RH,Cuenca-Estrella M.Invitroactivity of ASP2397 against Aspergillus isolates with or without acquired azole resistance mechanisms[J].Antimicrob Agents Chemother,2015,60(1):532-536.
[86] Nakamura I,Kanasaki R,Yoshikawa K,et al.Discovery of a new antifungal agent ASP2397 using a silkworm model ofAspergillusfumigatusinfection[J].J Antibiot (Tokyo),2017,70(1):41-44.
[87] Nakamura I,Yoshimura S,Masaki T,et al.ASP2397: a novel antifungal agent produced by Acremonium persicinum MF-347833[J].J Antibiot (Tokyo),2017,70(1):45-51.
猜你喜欢
西南农业学报(2020年8期)2020-12-10 01:50:02
中成药(2018年12期)2018-12-29 12:25:18
中成药(2018年3期)2018-05-07 13:34:18
现代检验医学杂志(2016年1期)2016-11-12 13:19:40
兽医导刊(2016年12期)2016-05-17 03:51:39
西南军医(2016年1期)2016-01-23 02:22:24
中国卫生标准管理(2015年5期)2016-01-14 05:17:06
山东医药(2015年13期)2016-01-12 00:39:39
中国医疗美容(2015年1期)2015-07-12 10:06:19
现代检验医学杂志(2015年2期)2015-02-06 02:01:11
