
高粱单核苷酸多态性(SNP)标记开发研究初报
2018-05-09 05:59籍贵苏杜瑞恒刘国庆侯升林李素英王金萍赵秀萍
华北农学报 2018年2期
籍贵苏,吕 芃 ,杜瑞恒 ,刘国庆 ,侯升林 ,马 雪 ,李素英 ,王金萍,赵秀萍
(1.河北省农林科学院 谷子研究所,河北省杂粮研究重点实验室,国家甜高粱改良分中心,河北 石家庄 050035;2.邯郸市永年区农牧业局,河北 邯郸 057050)
高粱抗旱耐瘠、耐盐碱,是当今世界尤为重要的能源作物和粮食作物。甜高粱生物产量高,其植体和茎秆丰富的糖汁均是生物乙醇生产的优质原料[1]。粒用高粱在我国历史上曾经是一个非常重要的济贫作物,而在缺水少雨的非洲,其仍然是人类食物的保障[2]。最近的研究证明,食用高粱对现代人类的健康,如减少炎症、肥胖、糖尿病以及高血脂等疾病上有重要作用[3]。
高粱基因组较小(750 Mb),被认为是二倍体模式化作物,同时也是能源植物如多倍体甘蔗和高生物量植物芒草类的参考植物[4-5]。高粱多态性标记的开发是图谱建立、数量性状和质量性状研究的基础,也是分子辅助育种和基因克隆的关键。高粱单核苷酸多肽性遍布于高粱的整个基因组,分布广、数量多,对其进行开发有助于扩大高粱的标记数量,为高密度图谱的建立、基因定位及基因开发意义重大。
最初高粱分子标记是用玉米和水稻等的探针或基因片段开发出来的,Hulbert等[6]经过对谷子、约翰逊草、甘蔗和珍珠粟等探针的比较,证明玉米探针最适合高粱PCR扩增。Berhan等[7]利用玉米的基因片段开发了96个标记,并用此构建了较早的高粱遗传图谱。随后较多的RFLP、AFLP等标记开发出来,对高粱多态性鉴定、图谱构建、性状鉴别等起到了重要的作用。
Peng等[8]用323个RFLP标记对高粱的一个重组自交系(RIL)群体137个个体进行分析,构建了10个连锁群,连锁群上的标记数为13~61个,长度为55~205 cM,总长度为1 347 cM,并把高粱与玉米的图谱的同源性作了详细的比较。Boivin等[9]整合了AFLP和RFLP构建的遗传图谱,形成了由443个位点总长1 899 cM的遗传图谱。
SSR具有稳定性和重复性好、分布广和费用低等特点,深受遗传学家的欢迎,尤其在遗传图谱的制作及连锁群的确认上有很大的优势。1997年Taramino等[10]开发了13个SSR标记,用其检测高粱的多态性及对高粱进行分析,把其中7个SSR标记标定在现有的高粱RFLP 图上。Kong等[11]通过高粱核基因文库合成了38个SSR引物,并用其对18个高粱品种和一个近等基因杂交群体进行扩增,把其中的31个SSR标记定位在高粱的连锁群上。2013年Kong等[5]又用203个SSR标记对由高粱与高粱近缘种(Sorhumbicolor×Sorghumpropinquum)杂交的F2和RIL群体进行分析和图谱构建,比较了141个等位基因位点在F2与RIL群体的10 条连锁群上的变化,提出RIL群体作图比F2群体更有优势。Wu等[12]用新开发的38个SSR标记连同以往的300多个标记对一个F2群体进行分析,构建了16个连锁群,分布在10个染色体上,证实了这些标记与原公布的染色体的位置有惊人的相似。另外,SSR标记在数量性状的标定上也显现出了它的优势,Guan等[13]利用118个SSR标记对粒用高粱石红137×甜高粱L-甜杂交的F2和F2:3的186个单株群体进行分析,构建了连锁群,对高粱含糖量、茎秆鲜质量及茎秆糖含量进行了QTL定位分析。Han等[14]对SSR标记把高粱干湿茎秆基因标定在第6染色体上。Wang等[15]通过多环境的SSR标记分析,认为染色体上与生物产量相关的主要QTL与多个性状相关,并存在着上位性效应。
多种标记的综合利用,无疑对挖掘高粱遗传信息,建立图谱具有深远的意义。Tao等[16]用8个SSR和155个RFLP标记对120 个 F5RIL群体进行分析,构建了一个长度为1 400 cM 的高粱图谱,分为21个连锁群,定位了3个性状。Menz等[4]对BTx623× IS3620C的 RIL群体用2 926个标记构建了长度为1 713 cM的10条连锁群。Ramu 等[17]开发了基于基因组序列的物理图谱,包含7 013个SSR标记,125个保守内含子扫描引物标记和100个已知基因位置。Haussmann等[18]为了将这些标记统一起来,用AFLP、SSR、RFLP 和 RAPD 标记对2个RIL群体进行了图谱构建,并将2个图谱合并成一套图谱,合并后的图谱包含了339个标记,11个连锁群,总长度1 424 cM。
分子标记在高粱的数量性状定位和分子标记辅助育种上取得了一定的进展。Agrama等[19]对抗蚜虫基因进行了定位,Murray等[20-21]对高粱含糖量进行了定位。Amelework等[22]用SSR标记对高粱品种的配合力和杂种优势进行研究,李浩杰等[23]利用分子标记辅助育种对高粱籽粒的淀粉含量进行了遗传改良。
2009年Paterson等[24]完成了高粱的测序,为高粱的基因组分析、遗传研究及基因的发掘利用等奠定了基础。随后,基于基因序列的研究,Bouchet等[25]用SNP标记对高粱的抽穗期和株高进行了标定。以往的标记尽管在高粱研究上起到了巨大的贡献,但只是限于有限的标记和特定的位点,基因遍布于整个基因组的各个部位,突变也千变万化,并且在标记不到的区间就很难找到潜在的基因,有限的标记难以满足分子遗传研究和标记辅助育种的需求。高通量测序由于可直接读取DNA序列变异,准确性高,同时还可机械化操作等迅速得到应用,用于基因组研究的SLAF(Specific lengh amplified fragments)标记,具有标记数量众多、均匀分布及避开重复序列等特点[26]。截至目前,其在芝麻、谷子、大豆等作物上得到广泛利用[27-29],尤其在作物高密度遗传图谱构建及功能基因验证上应用较多,而在高粱上的应用报道较少。用这种全基因组全方位扫描来筛选性状相关的基因标记方法,开发高密度遗传标记对优异基因的发掘及在高粱育种上的应用意义重大。
本研究对甜高粱与粒用高粱杂交的亲本及F2遗传群体进行基因组酶切,然后用高通量测序对其全基因组进行分析,开发SLAF标签并编码亲本及F2群体的基因型获得有效的SNP标记,旨在为遗传图谱构建、有益基因发掘、基因克隆和功能基因研究奠定基础,为分子标记辅助筛选和优异基因的利用提供材料和依据,提高我国高粱的研究水平,促进我国高粱和生物质能的发展。
1 材料和方法
1.1 研究材料
供试材料为甜高粱品种科特尔与粒用高粱J204杂交的F2群体。 J204 为来自美国的一个材料(由J14859变异单株选育而来)。2012年将该群体的父母本各10株和136个单株个体点播种植,生育期间按正常的田间管理进行,成熟时进行单株考种和数据采集。
1.2 DNA提取及处理
1.2.1 试验方法 CTAB方法提取DNA:生长期间对每株的最上一片叶片取样,-80 ℃保存待DNA提取,研磨样品中加入850 μL的CTAB,65 ℃水浴40 min后加入等体积的氯仿∶异戊醇(24∶1),振荡并离心,取上清加入0.6~0.7倍体积的异丙醇(-20 ℃预冷),沉淀,再离心,沉淀用75%的乙醇清洗2次,晾干,然后溶于100 μL TE中。
1.2.2 对DNA进行酶切 根据参考序列的 GC 含量、重复序列情况和基因特点等信息,选用MseⅠ的酶切组合,插入片段为 380~410 bp,对各样品DNA进行酶切。酶切过程用基因组 DNA 500 ng,加入NEB(New England Biolabs,NEB) 缓冲液 41 μL,补水至 50 μL,然后在37 ℃水浴中处理 15 h。反应结束后,用QIAGEN 胶提取盒纯化,50 μL EB回溶。
1.2.3 样品测序前的处理 酶切产物进行5′末端修复、3′末端加 A,连接测序接头,桥式扩增便于将连接产物锚定在Flowcell上,用琼脂糖凝胶电泳进行片段大小选择,通过PCR扩增切胶产物,增大文库量,建好的文库用 Illumina HiSeqTM2000进行测序[29]。
1.3 信息分析
1.3.1 SLAF标记开发 对各个样品同一位点处的 DNA片段序列,经过 PCR 扩增、测序,得到多个相同或相似的序列,使用 Blast 比对软件(http://www.blast.net/),通过序列之间存在的SNP 或 Indel来确定多态性,通过序列相似性将同一位点的片段序列进行聚类,每一个位点确定为一个群,群中高深度片段即为潜在基因型,采用关键参数为:tileSize=10 ,连续比对碱基数,最短比对单位长度stepSize=6 ,最短比对单位间的间距minScore=45 。聚类后,数据再进行测序错误的纠正,以每一个群中准确的高深度(≥50)序列作为参考,将低深度序列比对到参考序列上,对错配的碱基(错配数<5)进行纠正。消除错误SNP[29]。
1.3.2 编码基因型 纠错后获得具有 1个或多个(二倍体中,小于或等于 4个)等位基因的群,定义为SNP标记,只有1个等位基因的标记为非多态性标记,有2~4个等位基因的标记为多态性标记。利用8种分离模式(ab × cd、ef× eg、hk ×hk、lm × ll、nn × np、aa × bb、ab × cc和cc × ab) 对多态性标记编码基因型[29]。
1.4 测定项目及方法
株高:从地上部植株基部到穗顶的长度。茎节数:除穗茎节外,地上部可见茎节的个数。茎粗:用卡尺测量的茎秆从下数第三节茎秆的粗度。穗茎长:从最上一节结到穗柄的长度。百粒质量:去净谷壳和柄的百粒饱满籽粒称重。粒色:籽粒表皮颜色。抽穗期:本试验的抽穗期记载是从播种到穗抽出包叶的持续天数单株挂牌,为计算方便,本研究统一减去43 d得到的数值。穗长:从穗柄到穗顶的长度。穗鲜质量:从穗柄切下的鲜穗称重。茎鲜质量:去除穗和叶片后的植株称重。茎汁量:用榨汁机将茎秆扎压3次后,称取茎汁液的质量。茎糖量:取茎秆汁液一滴置入手持测糖仪 PAL-1 (日本 ATAGO Co.公司生产)上,记载读数。
2 结果与分析
2.1 标记开发群体的亲本表型性状的差异分析
较大的亲本差异有助于发掘更多的多态性,在调查的12个性状中,除了茎粗、穗长和抽穗期3个性状外,其他性状在父母本之间均达到显著差异(t检测)(表1)。
表1列出了作图群体亲本和F2的平均值及标准误。可见,作图群体的亲本遗传差异明显,除茎粗(P=0.77)、穗长(P=0.71)和抽穗期(P=1.00)外,调查的其他9个性状均有显著差异(P≤0.05)。从F2的标准误和平均值可见,群体有较大的分离,变异范围较大,其中株高274.92 cm、茎节数12.52个、穗长22.20 cm、茎鲜质量307.49 g,均超过其高值亲本科特尔,而F2的茎粗1.90 cm、穗质量53.30 g,则超过亲本J204。 这些性状属超高亲遗传。穗茎长、茎汁量、糖锤度和百粒质量则属偏高亲遗传,抽穗期F2均值为11.28 d,低于其双亲,说明F2的分离单株平均抽穗比亲本早3 d,属超亲遗传。另外,籽粒颜色在双亲之间颜色的分离也较大,出现了黄和灰色的非亲本色遗传分离。所以,作为多态性标记的开发群体,该试验群体完全符合试验需求。
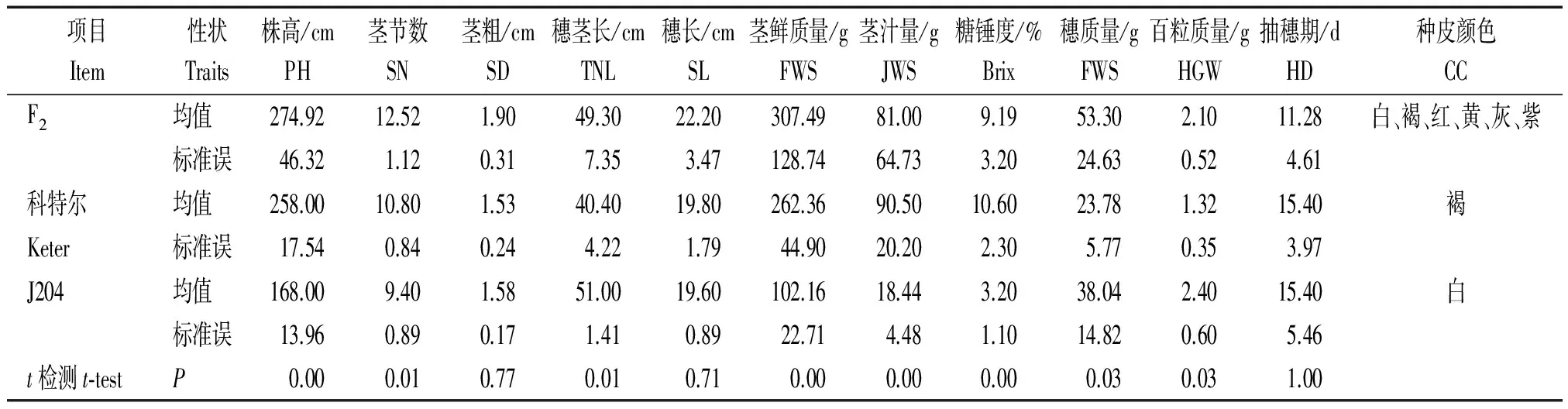
表1 亲本和F2群体12个性状的差异比较及检测Tab.1 Significance analysis and test for 12 traits in the parents and F2 population
注:PH. 株高;SN. 茎节数;SD. 茎粗;TNL. 穗茎长;SL. 穗长;FWS. 茎鲜质量;JWS. 茎汁量;Brix.糖锤度;FWS.穗质量;HGW. 百粒质量;HD.抽穗期;CC. 种皮颜色。F2种皮颜色分离:白/23,褐/68,红/22,黄/1,灰/3,紫/5 。
Note:PH. Plant hight;SN.Stem nods No.;SD.Stem diameter;TNL.Top nod length;SL.Spike length;FWS.Fresh weight of stem;JWS.Juice weight of stem;FWS.Fresh weight of spike;HGW.100-grain weight;HD.Heading date;CC.Coat color. The coat color distribution in the F2population is white/ 23,dark red/68,red/22,yellow/1,grey/3,purple/5.
2.2 测序结果统计
2.2.1 标记筛选基本情况 本次酶切片段长度为1~3 247 bp,总计302.6万个,多数片段分布在500 bp之内,占总片段的89.26%(图1)。
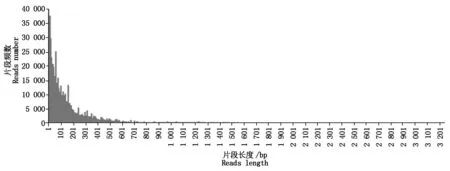
图1 酶切片段长度统计Fig.1 The frequency of reads lengths
本次测序采用双端测序,测序reads长度80 bp,各样品使用的reads 两端各40 bp长度进行统计(表2)。
由表2 可知,本次共开发的reads 数为43 528 021个,母本为2 598 472个,父本为3 134 524个,子代的reads数为205 083~454 258个,平均为290 732.5,GC平均含量为44.9%。
对上述测序reads进行原始数据评估、聚类和纠错等,开发到的亲本及各样品的SLAF标记数量以及测序深度如图2, 3所示。

表2 亲本和F2测序的reads 数和平均GC含量Tab.2 Reads numbers and GC contents from the parents and F2 individuals
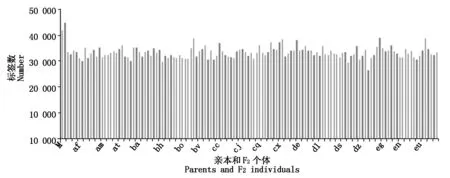
图中M和P为母本和父本,其他编号为130个F2个体。图3同。M and P refer to maternal and paternal parents and others refer to the F2 individuals.The same as Fig.3.
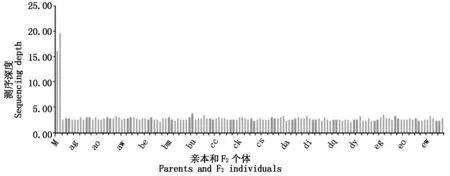
图3 亲本和F2个体的测序深度Fig.3 The sequencing depth in the parents and F2 individuals
可见,父母本的SLAFs 标签数分别44 895和42 100,测序深度在父本中为19.78 ,母本中为16.22。F2群体每个个体的 SLAF 标签为26 737~39 291,平均为 33 445.06,测序深度为2.24~3.72,平均为2.79。
通过对高深度片段的潜在基因型分析,共选定了 52 928 个较适宜的SLAF 标签,这些标签的测序总深度达到13 756 880。其中,多态性标签为 6 353 个(SNP 标记),占12.02%,测序深度累计为1 785 955,非多态性标签占46 575个,占88%,测序深度累计为11 970 925。
2.2.2 标记分型基本情况 根据F2群体基因型编码规则对 6 353 个多态性 SNP标记编码了基因型,其中,5 829 个标记成功分型,占多态性 SNP 标记的91.75%,不能成功分型的标记524个,占8.25%。成功分型的标记中,多数被编码的基因型为aa×bb,占87.37%,其次为lm×ll和nn×np,均为241个,hk×hk(143个),其他类型个数均很少,最低的是ab×cd,仅有2个(图4、表3)。
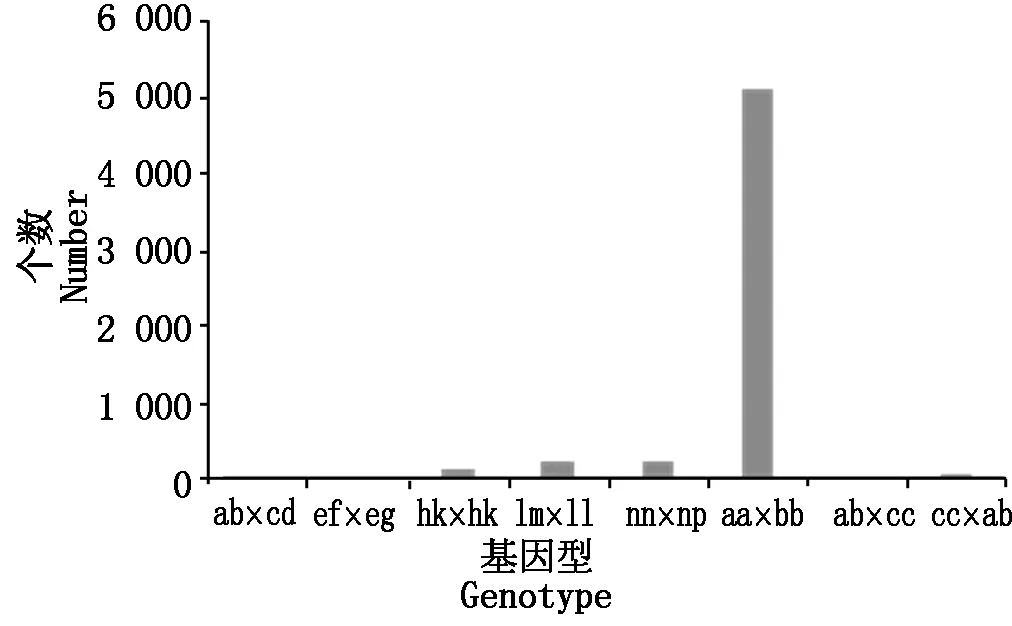
图4 成功分型标记各类型的频数Fig.4 The frequency of each genotype
表3列出了编码基因型的基本情况,524个不能分型的标记中包含了父母本缺失和重复序列等标记,占到99%。成功编码SNP标记中,父母本测序深度小于10 的标记占比最高,占45.7%(合计2 662个),其次非aa×bb类型的标记和SNP数多于3个的标记,分别占9.00%和4.90%。另外,还有可能为测序错误的标记和hk×hk标记中杂合度低于0.55的标记合计109个,剩余为有效的 SNP 标记2 246个。这2 246 个精选标记平均测序深度达到3.25,占有效标记百分比为100%,其F2个体的各样品完整度为85.99%~98.84%,平均为94.99%,可应用于进一步的图谱构建。
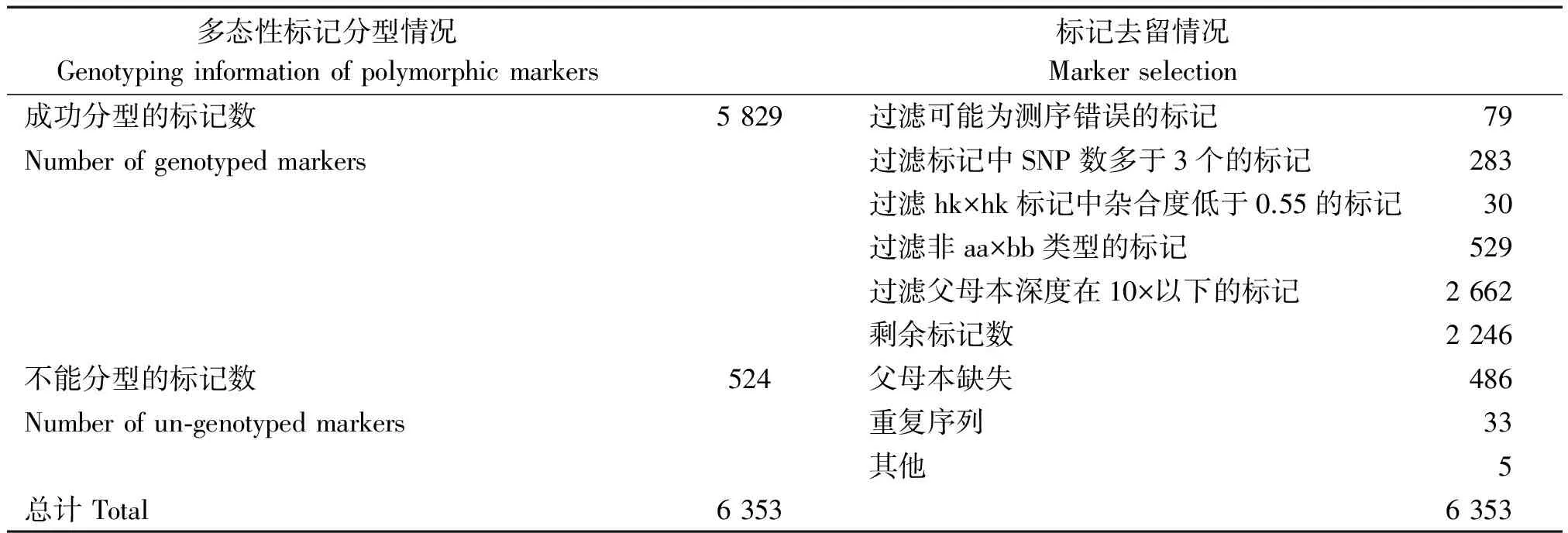
表3 多态性标记分型及去留基本情况一览表Tab.3 The genotyping information and marker selection of polymorphic markers
3 讨论与结论
本次研究利用了父母本在多性状上有较大差异的组合进行SLAF标签的开发,检测了株高、茎秆鲜质量、茎汁液鲜质量、含糖量等12个性状,其中,有9个性状在父母本之间差异显著。父母本差异越大,获得的多态性标记就越多,因此,本试验设计为更有效地发掘多态性标记奠定了基础。高通量测序方法的应用,检测了覆盖全基因组的碱基差异和变化,从而加大了标签开发的数量和质量[26],这在以往的标记和图谱研究中是无法达到的。该研究是从4 000多万reads中,通过聚类和纠错等筛选程序,获得了52 928 个SLAF 标签,并在这些标签中得到了6 353 个多态性SNP标记,测序深度累计为1 785 955。通过单核苷酸碱基变化获得的分子标记的数量之多,这在以往的研究方法和手段是难以达到的。
尽管SSR标记克服了AFLP和RFLP标记的随机性和不稳定性,但有限的数目难以满足基因组庞大数据和丰富基因变异的需求[29]。SLAF标签是据基因组的特性进行设计,更大程度地发现SNP、内含子(Indelible)和外显子(Extra)标记,增大了标记的数目,能更有效地标定有益基因,为基因克隆及在遗传育种上的应用奠定坚实的基础,并可据已知的序列设计引物用于基因组比较及分子标记辅助育种,同时作为模式作物可促进其他作物遗传研究的发展[5]。
该研究利用亲本遗传差异较大的材料进行杂交,获得性状变异丰富的F2群体,有利于多态性标记的开发,通过高通量测序和覆盖全基因组标记的检测,从43 528 021个测序片段中获得了 6 353 个多态性 SNP 标记,其中,5 829 个标记成功分型,在这些分型的标记中又精选出2 246个有效的 SNP 标记,可应用于进一步的图谱构建。
参考文献:
[1] Vinutha K S,Rayaprolu L,Yadagiri K,et al.Sweet sorghum research and development in India:status and prospects[J]. Sugar Tech,2014 16(2):133-143.
[2] Hadebe S T,Modi A T,Mabhaudhi T. Drought tolerance and water use of cereal crops:a focus on sorghum as a food security crop in Sub-Saharan Africa[J]. Journal of Agronomy and Crop Science,2017,203(3):177-191.
[3] Cardoso L D,Pinheiro S S,Martino H S D,et al. Sorghum (SorghumbicolorL.):nutrients,bioactive compounds,and potential impact on human health[J]. Critical Reviews in Food Science and Nutrition,2017,57(2):372-390.
[4] Menz M,Klein R R,Mullet J E,et al. A high-density genetic map ofSorghumbicolor(L.)Moench based on 2926 AFLP,RFLP and SSR markers[J]. Plant Molecular Biology,2002,48(5/6):483-499.
[5] Kong W Q,Jin H Z,Woodfin C,et al.Genetic analysis of recombinant inbred lines forSorhumbicolor×Sorghumpropinquum[J].G3 Genes/Genomes/Genetics,2013(3):101-108.
[6] Hulbert S H,Richter T E,Axtell J D,et al. Genetic mapping and characterization of sorghum-related crops by means of maize DNA probes[J]. Proc Natl Acad Sci USA,1990,87(11):4251-4255.
[7] Berhan M A,Hulbert S H,Butler L G,et al. Structure and evolution of the genomes ofSorghumbicolorandZeamays[J]. Theor Appl Genet,1993,86(5):598-604.
[8] Peng Y,Schertz K F,Cartinhour S,et al. Comparative genome mapping ofSorghumbicolor(L.) Moench using an RFLP map constructed in a population of recombinant inbred lines[J].Plant Breeding,1999,118(3):225-235.
[9] Boivin K,Deu M,Rami J F,et al. Towards a saturated sorghum map using RFLP and AFLP markers[J]. Theoretical and Applied Genetics,1999,98(2):320-328.
[10] Taramino G,Tarchini R,Ferrario S,et al. Characterisation and mapping of simple sequence repeats (SSRs) inSorghumbicolor[J]. Theor Appl Genet,1997,95(1-2):66-72.
[11] Kong L,Dong J,Hart G E. Characteristics,linkage-map positions ,and allelic differentiation ofSorghumbicolor(L.) Moench DNA simple-sequence repeats (SSRs)[J]. Theor Appl Genet,2000,101(3):438-448.
[12] Wu Y Q,Huang Y H. An SSR genetic map ofSorghumbicolor(L.) Moench and its comparison to a published genetic map[J]. Genome,2007,50(1):84-89.
[13] Guan Y A,Wang H L,Qin L ,et al. QTL mapping of bio-energy related traits in Sorghum[J].Euphytica,2011,182:431.
[14] Han Y C,Lv P,Hou S L,et al. Combining next generation sequencing with bulked segregant analysis to fine map a stem moisture locus in sorghum(SorghumbiocolorL.moench)[J]. PLoS One,2015,10(5):e127065.
[15] Wang H L,Zhang H W,Du R H,et al. Identification and validation of QTLs controlling multiple traits in sorghum[J]. Crop and Pasture Science,2016,67(2):193-203.
[16] Tao Y Z,Jordan D R,Henzell R G,et al. Construction of genetic map in aa sorghum recombinant inbred line using probes from different sources and its conparision with other sorghum maps[J]. Australian Journal of Agricultural Research,1998,49:729-736.
[17] Ramu P,Kassahun B,Senthilvel S,et al. Exploiting rice-sorghum synteny for targeted development of EST-SSRs to enrich the sorghum genetic linkage map[J]. Theoretical and Applied Genetics,2009,119(7):1193-1204.
[18] Haussmann B I G,Hess D E,Seetharama N,et al. Construction of a combined sorghum linkage map from two recombinant inbred populations using AFLPs,SSR,RFLP,and RAPD markers,and comparison with other sorghum maps[J]. Theor Appl Genet,2002,105(4):629-637.
[19] Agrama H A,Widle G E,Reese J C ,et al.Genetic mapping of QTLs associated with greenbug resistance Sorghum bicolor and tolerance inSorghumbicolor[J]. Theor Appl Genet,2002,104(9):1373-1378.
[20] Murray S C,Rooney W L,Hamblin M T. et al. Sweet Sorghum genetic diversity and association mapping for brix and height[J]. Plant Genome,2009,2(1):48-62.
[21] Lv P,Ji G S,Han Y C,et al.Association analysis of sugar yield-related traits in sorghumSorghumbicolor(L.)[J]. Euphytica,2013,193(3):419-431.
[22] Amelework B,Shimelis H,Laing M. Genetic variation in sorghum as revealed by phenotypic and SSR markers:implications for combining ability and heterosis for grain yield[J]. Plant Genetic Resources-Characterization and Utilization,2017,15(4):335-347.
[23] 李浩杰,李 平,高方远,等. SSR标记辅助选择改良冈46B直链淀粉含量的研究[J]. 作物学报,2004,30(11):1159-1163.
[24] Paterson A H,Bowers J,Bruggmann R,et al. TheSorghumbicolorgenome and the diversification of grasses[J]. Nature,2009,457:551-556.
[25] Bouchet S,Olatoye M O,Marla S R,et al. Increased power to dissect adaptive traits in global sorghum diversity using a nested association mapping population[J].Genetic,2017,206(2):573-585.
[26] Sun X W,Liu D Y,Zhang X F,et al. SLAF-seq:an efficient method of Large-Scale De Novo SNP discovery and genotyping using High-Throughput sequencing[J]. PLoS One,2013,8(3):e58700.
[27] Zhang Y X,Wang L H,Xin H G,et al. Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing[J]. BMC Plant Biology,2013,13:141.
[28] Bai H,Cao Y H,Quan J Z,et al. Identifying the genome-wide sequence variations and developing new molecular markers for genetics research by re-sequencing a Landrace cultivar of foxtail millet[J]. PLoS One,2013,8(9):e73514.
[29] Li B,Tan L,Zhang J Y,et al. Constuction of high density genetic map based on large-scale markers developed by specific length amplified fragmeng sequencing(Slaf-seq)and its application to QTL analysis for isoflavone content inGlycinemax[J]. BMC Genomics,2014,15(11):e1086.
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
古今农业(2022年2期)2022-08-15
西南农业学报(2022年5期)2022-06-06
昆明医科大学学报(2022年2期)2022-03-29
青年文学家(2022年1期)2022-03-11
今日农业(2021年20期)2021-11-26
昆明医科大学学报(2021年6期)2021-07-31
昆明医科大学学报(2021年3期)2021-07-22
中国糖料(2021年3期)2021-07-13
热带农业科技(2019年1期)2019-01-14
