
煤液化残渣中硫的氧化规律
2018-04-25 01:24:14常卫科袁桂梅陈胜利张胜振
中国粉体技术 2018年2期
常卫科,徐 洁,孙 伟,袁桂梅,陈胜利,张胜振
(1.中国石油大学(北京) 重质油国家重点实验室,北京 102249;2.北京低碳清洁能源研究所,北京 102211)
煤液化残渣是煤在高温、高压条件下直接催化加氢的液化产物经常减压蒸馏后,不能蒸馏出来的物质。其中含有高沸点有机物、未转化的煤粉、煤中的无机矿物和加入的加氢催化剂(通常是铁)。煤液化残渣产量高达原煤使用量的20%~30%[1]。由于在煤液化过程中主要使用铁基催化剂(硫铁矿),并加入硫单质作为助剂[2],在整个工艺过程中硫在残渣中富集,并最后以磁黄铁矿和黄铁矿的形式存在于液化残渣中,导致液化残渣的硫含量超过2.5%(质量分数,下同),高于原煤中硫含量[3-4]。
目前,煤直接液化残渣主要利用途径有加氢液化、干馏(热解、焦化)、气化制氢、直接燃烧等[5-8]。高硫含量导致现存的煤液化残渣利用方式存在费用高、残渣利用效率低、环境危害大等缺点,尤其是直接燃烧造成严重的环境污染。煤液化残渣的软化点在130℃以上[9],高于普通石油沥青,并且与道路集料粘附性能极强,煤液化残渣可直接作为石油沥青改性剂,改性后沥青可满足美国 ASTM和英国BSI的标准要求。但残渣中的硫对沥青性质有重要影响,若要更好地将煤液化残渣应用在道路沥青领域,需要对研究沥青热拌状态下硫的变化规律。故此研究了200~300℃下,煤液化残渣中的硫在空气中的氧化规律和动力学,为液化残渣的有效利用提供参考。
1 实验
1.1 煤液化残渣的分离预处理
称取10 g煤液化残渣(CDLR)于锥形瓶中,加入8倍质量的四氢呋喃,在50℃下充分溶解2 h,抽滤,并用50℃四氢呋喃洗涤滤饼至滤液基本无色;滤饼放于真空干燥箱中80℃下真空干燥2 h,得到四氢呋喃不溶物(TFHI)。滤液放于通风橱中,使溶液中四氢呋喃挥发除去,剩余部分为四氢呋喃可溶物(THFS)。
测定CDLR、TFHI和TFHS的质量和硫含量,结果如表1所示。
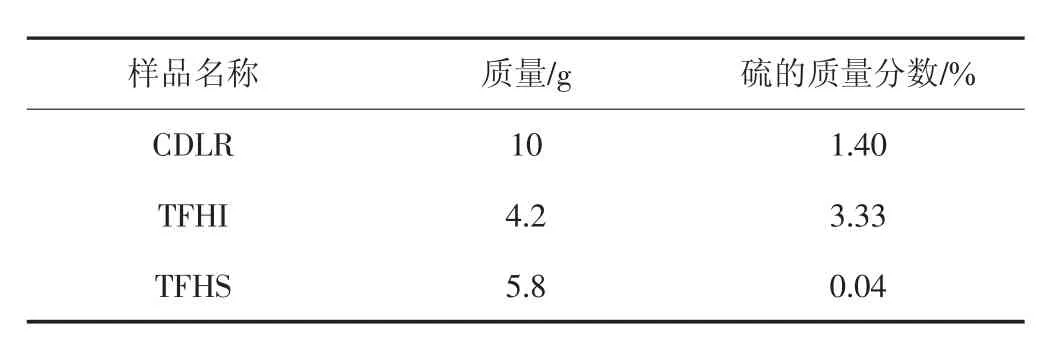
表1 样品质量及硫含量Tab.1 Sulfur content of several samples
由表1可知,THFI含硫量远高于THFS,煤液化残渣中的硫主要存在于THFI中。
1.2 氧化反应
取3 g四氢呋喃不溶物与15 g石英砂(粒径0.85~2.00 mm)混合,装入管式反应器中,氮气保护下升温至实验温度。然后将N2切换成干空气(压缩空气)进行氧化反应。空气体积流量为200 mL/min,分别反应 15、30、60、120、180 min 后将空气切换成氮气,并将温度降至室温后取出样品。氧化后的样品经研磨后,分析样品中各形态硫含量。
湿空气氧化时,空气进入反应器前进行加湿,含水量约2.7%(质量分数),其他步骤与干空气氧化实验相同。
1.3 样品不同形态硫含量测定
THFI中硫可分为硫酸盐(SO42-)硫 Sa、磁黄铁矿(FeS)硫 St、黄铁矿(FeS2)硫 Se 和硝酸不溶物硫 SNI。样品不同形态硫含量分析过程如图1所示。
实验的主要仪器有:高频红外碳硫仪(HIR-944B,无锡市高速分析仪器有限公司)、X射线光电子衍射仪(XPS,OXFORD Link ISIS300,英国)、紫外荧光定硫仪(SY-2000S,泰州市姜堰分析仪器厂,燃烧室温度为1 000℃)等。
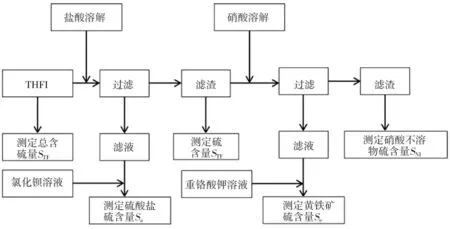
图1 样品不同形态硫含量分析过程Fig.1 Procedures of sulfur content analysis
1.3.1 样品总含硫量的测定
采用高频红外碳硫仪测定样品总含硫量STF。
1.3.2 硫酸盐硫的测定
采用国标GB/T215—2003中的标准方法测定样品中硫酸盐硫(SO42-)含量。测定过程:取1.0 g左右的样品,加入10 mL的稀盐酸(5 mol/L),微沸30 min,样品中硫酸根被浸出,FeS与HCl反应生成H2S气体,FeS2和有机硫仍固定在固体中,过滤后,向滤液中滴加过量的氯化钡溶液,生成硫酸钡沉淀,经过滤、干燥、焙烧等过程,根据硫酸钡的质量计算煤中硫酸盐硫含量Sa,计算公式为
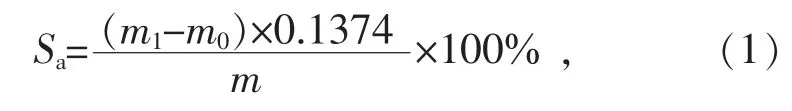
式中:m为样品质量,g;m0、m1分别空白、样品测定的硫酸钡质量,g;0.1374为硫在硫酸钡中的质量占比。
1.3.3 黄铁矿硫的测定
采用国标GB/T 215—2003中的标准方法测定样品中黄铁矿硫(FeS2)含量。在稀盐酸浸取后的固体样品中加入定量的稀硝酸,微沸30 min,样品中FeS2与稀硝酸(加2 mL过氧化氢溶液)反应生成Fe3+和H2S气体。用二氧化锡将Fe3+还原为Fe2+。然后用重铬酸钾标准溶液滴定Fe2+,根据重铬酸钾标准溶液的消耗量确定铁的质量,再以铁的质量计算样品中硫化铁(黄铁矿)硫含量Se,计算公式为
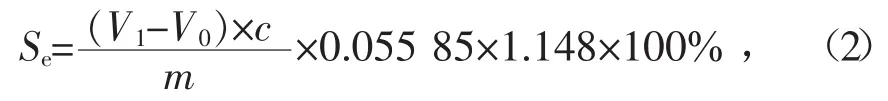
式中:V1、V0分别是实验测定和空白测定时铬酸钾标准溶液用量,mL;c为重铬酸钾标准溶液浓度,mol/L;0.05585为铁的毫摩尔质量,g/mmol;1.148为由铁换算成硫化铁硫的系数;m为样品质量,g。
1.3.4 磁黄铁矿硫的测定
盐酸浸取THFI过程中,磁黄铁矿硫(FeS)和硫酸盐硫进入溶液中。磁黄铁矿硫So含量采用差减法计算,计算公式为

式中:STF为 THFI总含硫量(质量分数),%;SHCl为盐酸浸取后THFI含硫量(质量分数),%。
1.3.5 硝酸不容物硫的测定
硝酸浸取后的THFI中的硫为硝酸不溶物硫(SNI),其含量采用高频红外碳硫仪测定硝酸浸。
2 结果与讨论
2.1 残渣样品各形态硫分布
对THFI进行硫分析,表2为氧化前THFI固体中不同形态硫的质量分数及占总硫百分比。由表2可知,THFI中So占到总含硫量的74.17%,液化残渣中硫主要以磁黄铁矿的形式存在,Se、Sa和SNI含量较少。
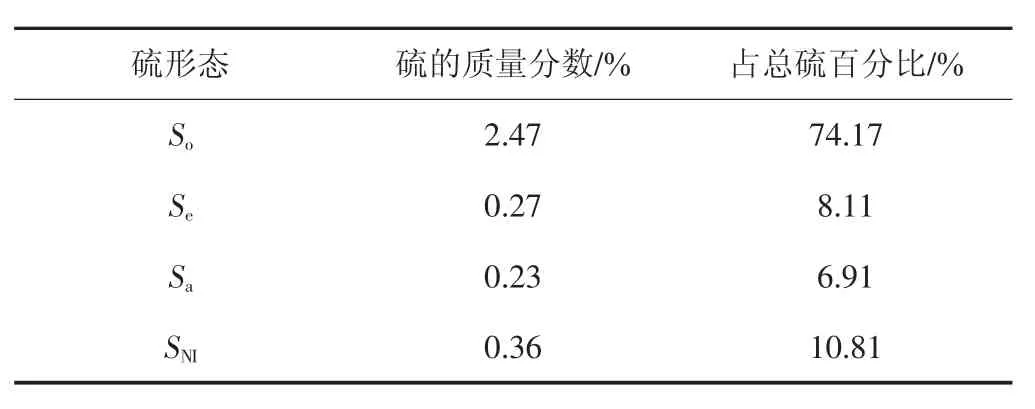
表2 氧化前THFI各样品硫的质量分数及占总硫百分比Tab.2 Sulfur contents in different types of sulfur-containing components in the THFIs
2.2 干空气氧化THFI
2.1.1 THFI总含硫量变化规律
图2为干空气氧化时间对THFI含硫量的影响。由图可知,各温度条件下,THFI总硫含量随着氧化反应时间的增加而减少,且氧化温度越高硫含量下降越明显,即THFI含硫量下降速率,230℃<250℃<270℃。
氧化前后反应管出口处石英砂照片见图3。在氧化过程中发现,反应管出口处填充的石英砂表面附着了一层黄色物质,对其进行X射线光电子能谱(XPS)分析,结果如图4所示。与标准谱图对比后可知,石英砂表面附着物中的含硫物质主要是单质硫。

图3 氧化前后反应管出口处石英砂照片Fig.3 Photo of quartz sand in exit side of reactor before oxidation and after oxidation
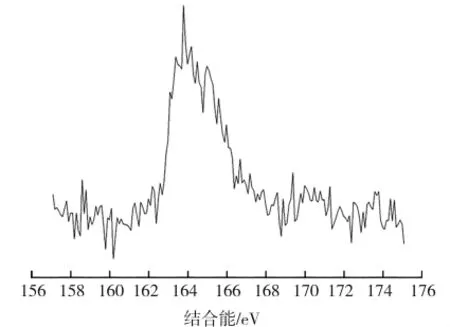
图4 反应管出口处石英砂表面附着物XPS谱图Fig.4 Analysis of sulfur valence in species adsorbed on surface of quartz sand located at exit side of reaction tube(XPS)
根据文献报道[10-13],磁黄铁矿和黄铁矿易与空气中氧气发生氧化反应,常温下反应产物主要是硫酸盐,随着温度的升高会有单质硫生成。单质硫在试验温度下升华被流动空气带到反应管出口处,由于反应管出口处的温度低(约为55℃),气体中的硫单质凝华并附着在反应管出口处的石英砂上,这是反应后样品总含硫量降低的原因。
2.1.2 THFI各形态硫含量变化规律
图5—8分别是干空气氧化时间对THFI样品中So、Se、Sa和 SNI含量的影响。
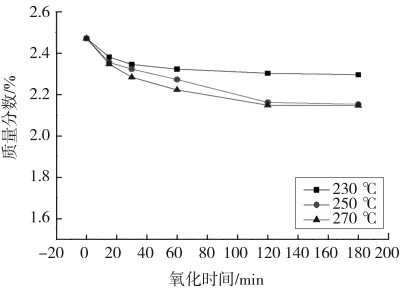
图5 干空气氧化THFI过程中氧化时间对So的影响Fig.5 Effect of oxidation time on Soof THFI in dry air
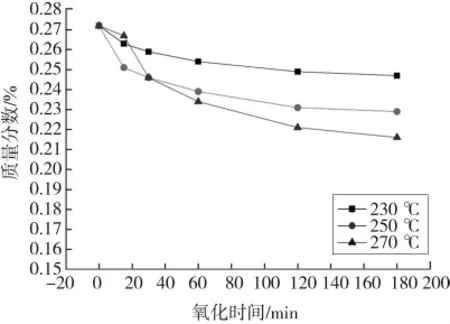
图6 干空气氧化THFI过程中氧化时间对Se的影响Fig.6 Effect of oxidation time on Seof THFI in dry air
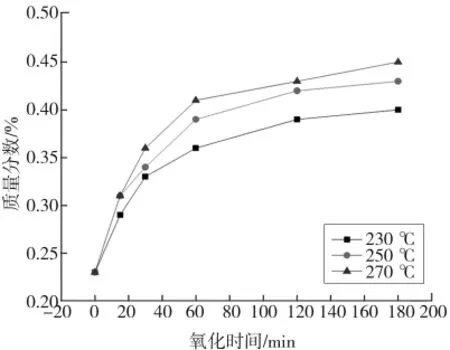
图7 干空气氧化THFI过程中氧化时间对Sa的影响Fig.7 Effect of oxidation time on Saof THFI in dry air
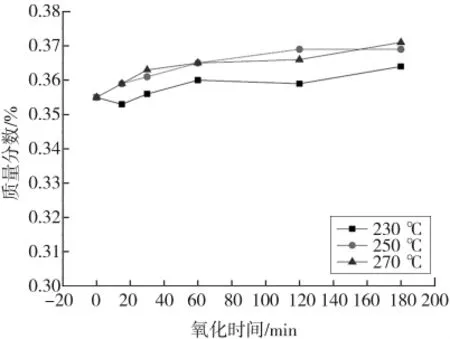
图8 干空气氧化THFI过程中氧化时间对SNI的影响Fig.8 Effect of oxidation time on SNIof THFI in dry air
由图5—图8可知,各温度条件下So和Se含量随着氧化反应的进行而减少,120 min后基本不再变化;Sa和SNI含量随着氧化反应的进行而增加,且增加速率逐渐降低。
2.2 湿空气氧化THFI
为了探究空气中水对煤液化残渣中硫的氧化是否影响,利用湿空气氧化THFI样品,即在空气进入氧化反应器之前,对其进行加湿处理,其他条件与干空气氧化THFI样品一致。
2.2.1 THFI总含硫量变化规律
图9为湿空气氧化时间对THFI样品硫含量的影响。由图可知,不同温度下,THFI总硫含量随着氧化反应时间的增加没有发生明显的变化。同时发现,氧化过程中反应管出口处的石英砂没有黄色物质附着,说明在湿空气氧化过程中没有单质硫生成。采用紫外荧光定硫仪对反应管尾气进行含硫量分析,同样没有发现气相中有硫化物的存在。
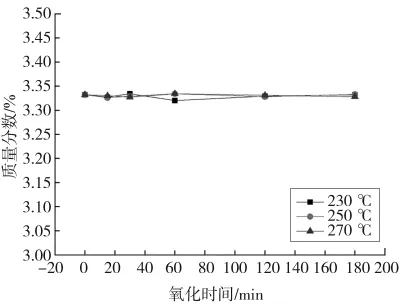
图9 湿空气氧化时间对THFI含硫量的影响Fig.9 Change of sulfur content of THFI with oxidation time in wet air
2.2.2 THFI总含硫量变化规律
图10—13分别表示湿空气氧化时间对THFI样品中So、Se、Sa和SNI含量的影响。由图可以看出,反应条件下,THFI样品中So和Se含量随着氧化反应的进行而逐步降低,温度越高变化速率越大,120 min后基本不再变化;Sa含量和SNI含量随着氧化的进行而增加,增加速率逐渐降低。
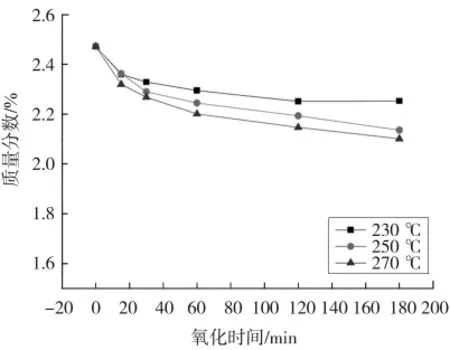
图10 湿空气氧化THFI过程中氧化时间对So的影响Fig.10 Effect of oxidation time on Soof THFI in wet air
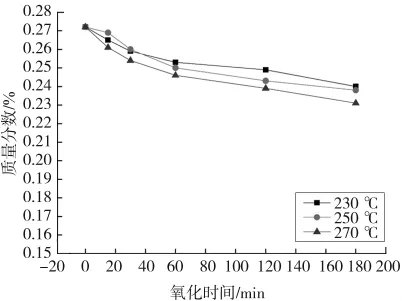
图11 湿空气氧化THFI过程中氧化时间对Se的影响Fig.11 Effect of oxidation time on Seof THFI in wet air
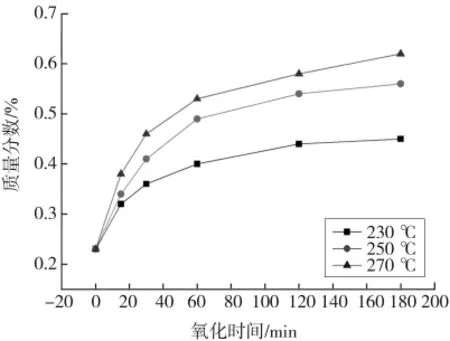
图12 湿空气氧化THFI过程中氧化时间对Sa的影响Fig.12 Effect of oxidation time on Saof THFI in wet air
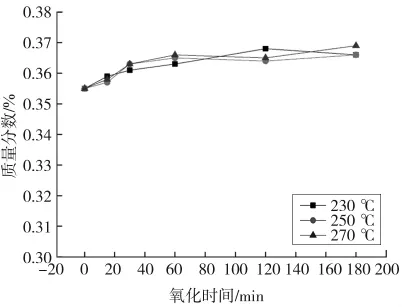
图13 湿空气氧化THFI过程中氧化时间对SNI的影响Fig.13 Effect of oxidation time on SNIof THFI in wet air
图14为相同条件下干、湿空气氧化THFI样品中Sa含量随氧化时间的变化趋势对比图。由图可知,湿空气氧化样品硫酸盐硫增加量大于相同条件下干空气氧化样品硫酸盐硫增加量。据相关研究[11、14-18],单质硫是硫铁矿氧化的重要中间产物,水可以改变硫铁矿的氧化历程,加速中间产物(单质硫)进一步氧化生成硫酸根。
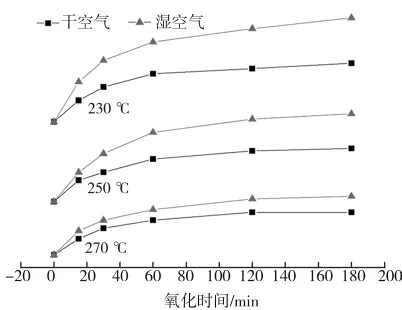
图14 干、湿空气氧化样品硫酸盐硫含量随氧化时间变化对比Fig.14 Comparison of sulfur content of Sain THFIs oxided in dry air and wet air at differnent temperatures for different reaction times
2.3 干空气氧化THFI动力学模型
煤液化残渣在空气氧化过程中的反应物主要是磁黄铁矿和黄铁矿,由于煤液化残渣中黄铁矿的含量只有磁黄铁矿含量的1/10,且磁黄铁矿比黄铁矿更易氧化[19],所以可认为反应物为磁黄铁矿,即FeS。干空气下的反应的产物硫酸盐和硫单质据此可推得不同条件下的反应方程式为
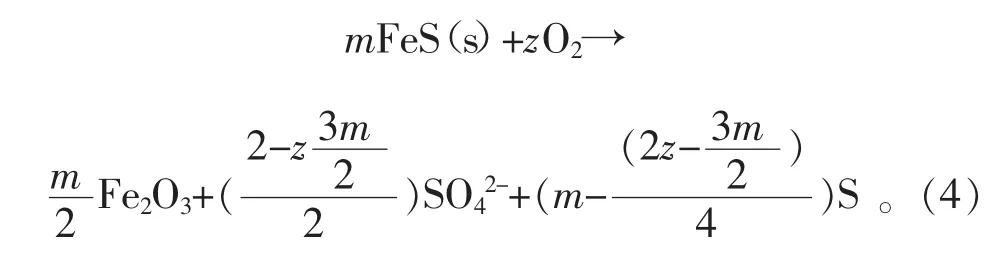
比较多种动力学模型(包括颗粒表面反应模型、颗粒体相反应模型、灰层扩散控制模型等),发现氧气通过颗粒灰层扩散控制模型最符合实验事实。为了进一步验证氧化过程是氧在灰层中扩散控制,取270℃下干空气氧化180 min后的样品进行充分研磨,压片后研磨成颗粒(0.250~0.425 mm);在270℃下对得到的固体颗粒进行不同时间的干空气氧化实验,方法过程与初次氧化过程相同。图15、16分别为研磨后再氧化THFI样品中So和Sa含量与氧化时间的关系曲线。
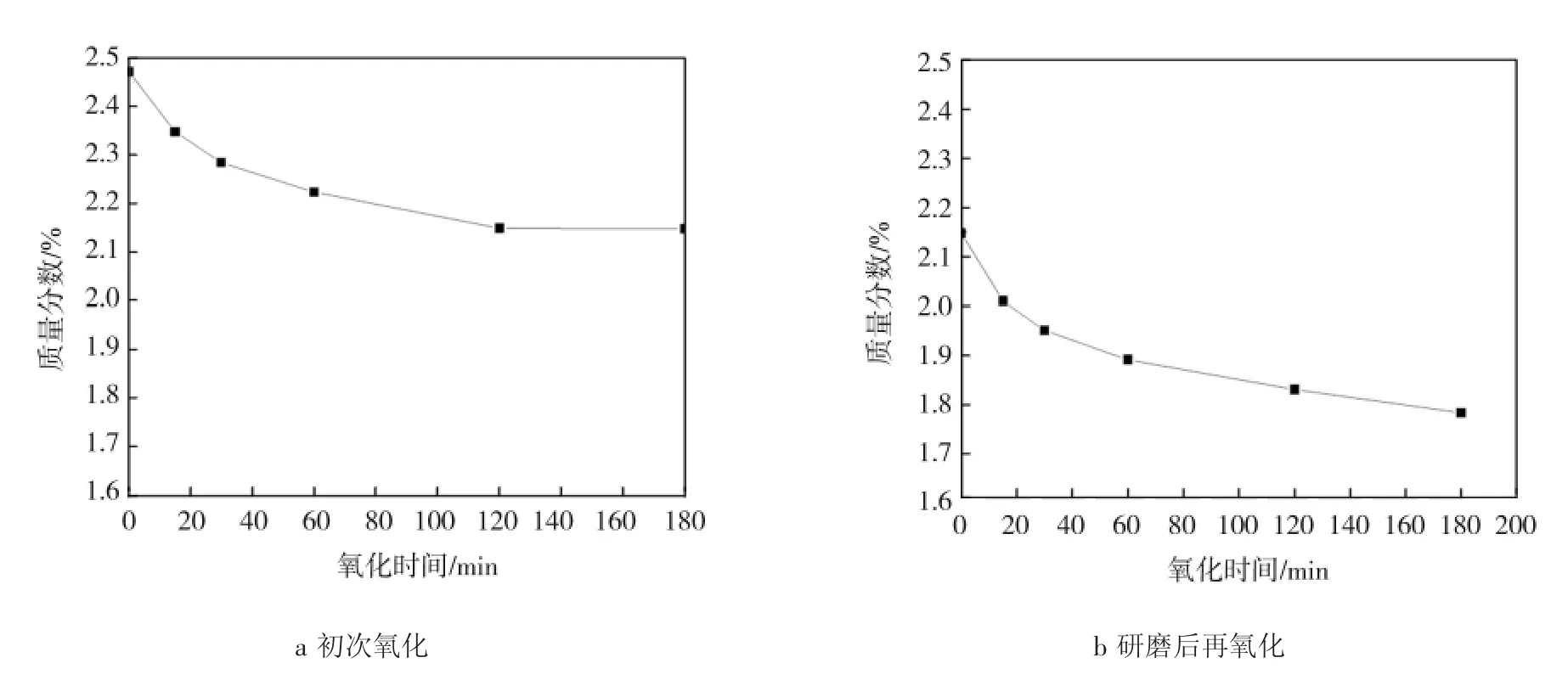
图15 样品初次氧化样品和研磨后再氧化样品中So与氧化时间的关系Fig.15 Relationship between oxidation time and content of Soin initial oxidated sample and subsequent oxidation sample
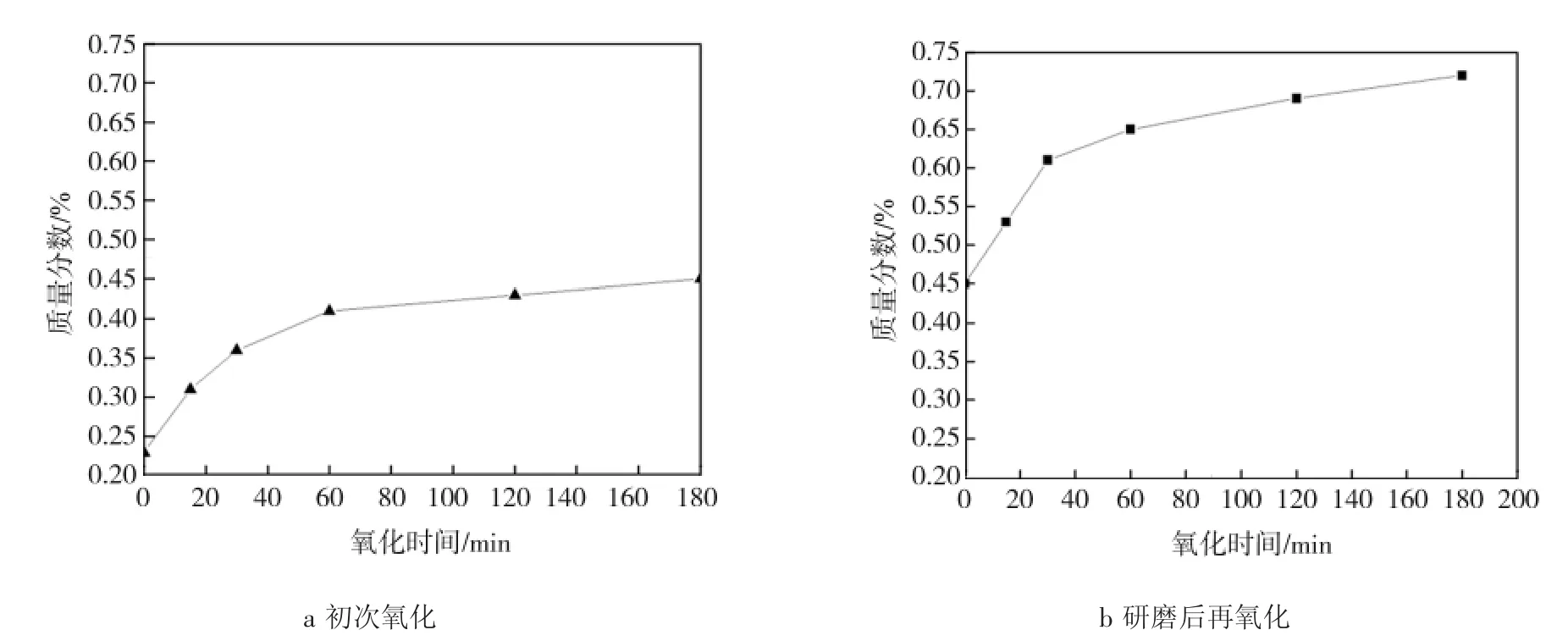
图16 样品初次氧化样品和研磨后再氧化样品中Sa含量与氧化时间的关系Fig.16 Relationship between toxidation time and content of Sain initial oxidated sample and subsequent oxidation sample
由图15、16可知,THFI经干空气 270℃氧化180 min后,磁黄铁矿的硫含量基本不再降低,氧化产物硫酸盐含量也基本不变;经研磨后再次氧化,磁黄铁矿硫含量又开始降低,其氧化产物硫酸盐含量又迅速上升,之后变化趋于平缓。这充分证明,实验条件下,氧化过程由磁黄铁矿颗粒上覆盖的硫酸盐产物层中的氧扩散控制。第1次氧化后,磁黄铁矿表面覆盖了一层硫酸盐膜,减缓或阻止了磁黄铁矿进一步氧化。经过研磨后,硫酸盐灰层从磁黄铁矿表面脱落,暴露的磁黄铁矿再一次被氧化。但当磁黄铁矿表面再次被硫酸盐灰层覆盖后,氧化速度再次减慢。样品在氧化过程中,FeS由外至内逐步消耗,且会在表面留下一层灰层,空气中的氧气必须穿过灰层才能与FeS继续反应。灰层扩散控制模型示意图见图17,CA0、CAS分别为气流主体和FeS表面的氧气浓度。
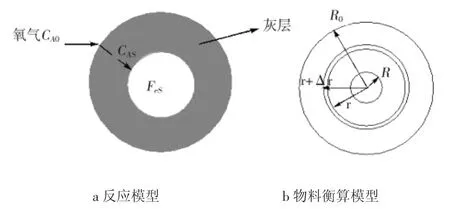
图17 灰层扩散控制模型示意图Fig.17 Diffusion-controlled model of oxygen through surface ash layer of THFI
如图17b所示,R0为颗粒粒径,R为未反应颗粒粒径,r为氧气通过某截面距颗粒球心的距离。根据灰层扩散控制模型[20]可推得:
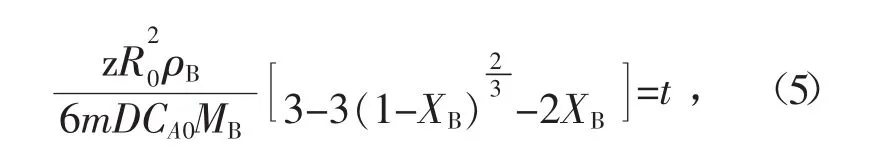
式中:D为扩散系数,m2/s;ρB为FeS颗粒密度,kg/m3;MB为FeS相对分子量,g/mol;XB为FeS转化率。令
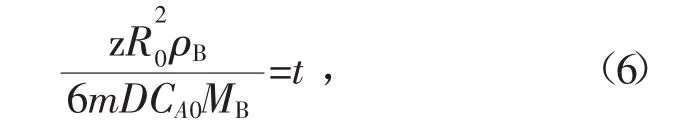
则式(1)可写为
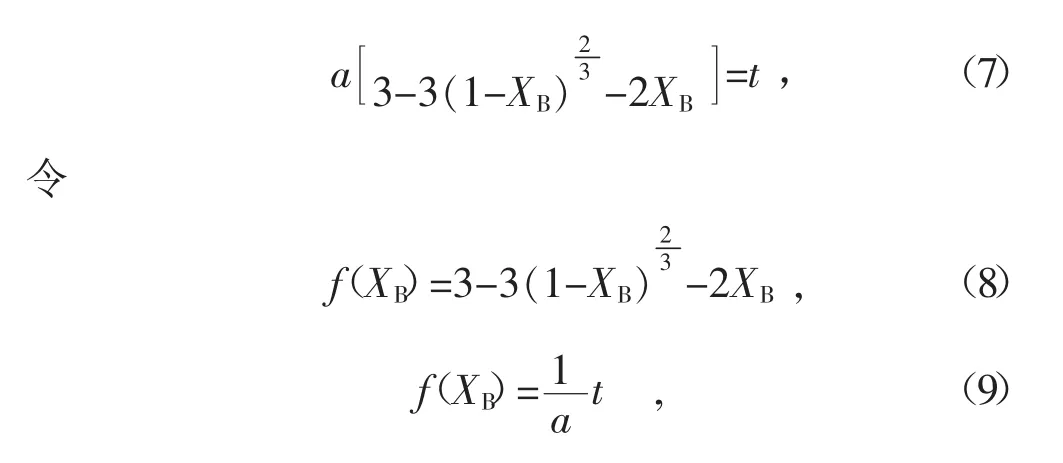
以f(XB)为纵坐标,反应时间t为横坐标作图,若f(XB)与氧化时间t为线性关系,则灰层扩散控制模型符合空气氧化煤液化残渣四氢呋喃不溶物固体。图18为230、250、270℃下干空气氧化样品f(XB)与氧化时间t的关系曲线,由图可知,f(XB)与反应时间t为线性关系。所以,实验条件下,氧化过程由磁黄铁矿颗粒上覆盖的硫酸盐产物层中的氧扩散控制。
目前已有文献 [21-24]将有效扩散系数采用Arrhenius关系式进行描述。那么有效扩散系数与扩散活化能的关系便可描述为
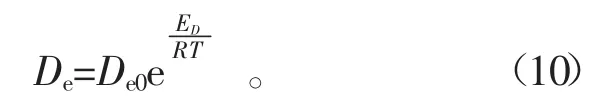
式中:De、De0分别为有效扩散系数和指前因子,m2·s-1;ED为扩散活化能,kJ·mol-1;T 为扩散温度,K;R 为气体状态常数,R=8.314 J·mol-1·K-1。
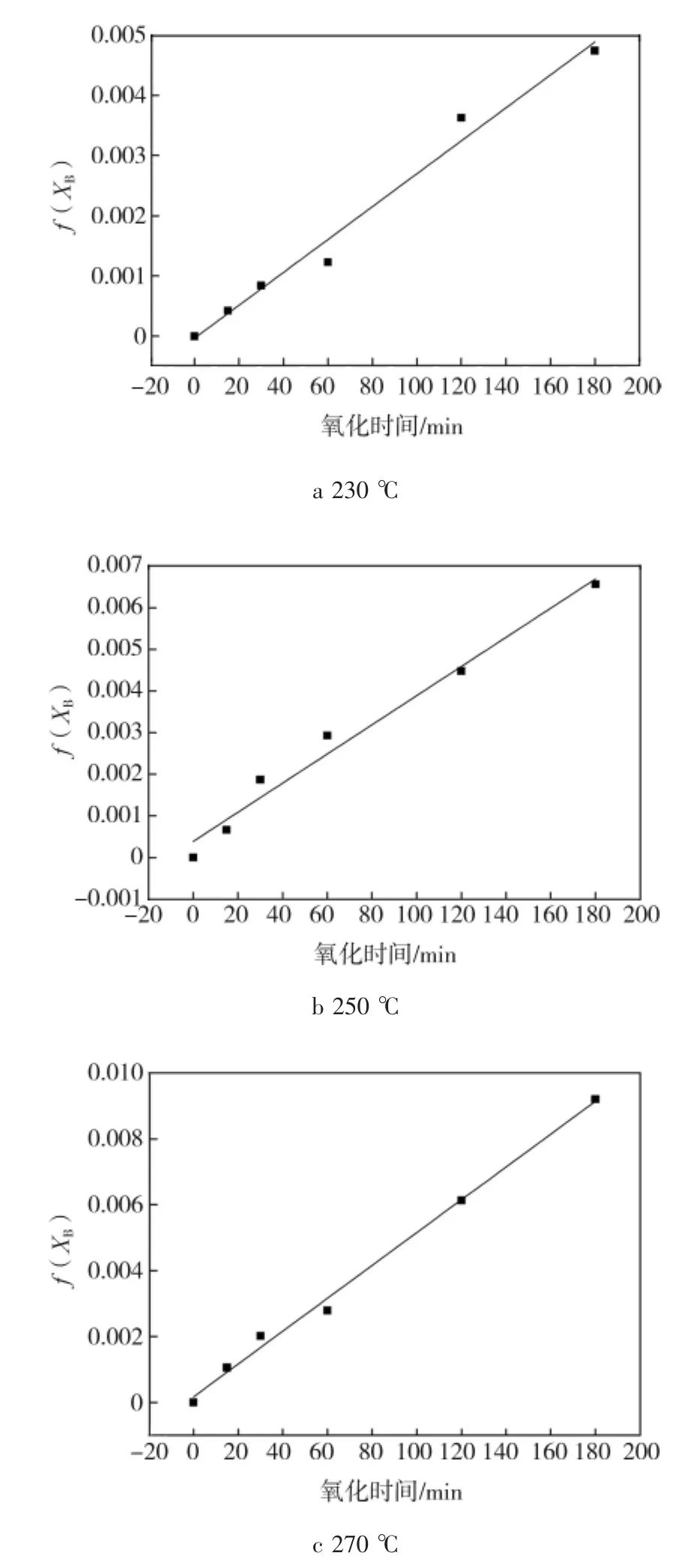
图18 f(XB)与氧化时间t的关系曲线Fig.18Variation of f(XB)vs oxidation time
表3 干空气氧化反应模型中值Tab.3 of dry air oxidation reaction model

表3 干空气氧化反应模型中值Tab.3 of dry air oxidation reaction model
反应温度/℃ 230 250 270 1 a/s-1 2.69×10-5 3.36×10-5 4.19×10-5
3 结论
煤液化残渣中的硫主要以磁黄铁矿形式存在,占总含硫量的74.2%;在实验条件下,干空气氧化四氢呋喃不溶物,反应生成硫酸盐和单质硫,单质硫实验温度下,升华被流动空气带出反应系统,样品的总含硫量减少;空气中的水对样品氧化过程中硫酸盐的生成有促进的作用,湿空气氧化残渣样品,无单质硫生成。在实验条件下,氧化过程由磁黄铁矿颗粒上覆盖的硫酸盐产物层中的氧扩散控制,扩散活化能为26.25 kJ/mol。
参考文献(References):
[1]黄传峰,韩磊,王孟艳,等.煤加氢液化残渣的性质及应用研究进展[J].现代化工,2016(6):19-23.
[2]吴琼,高宇龙,刘柯澜.神华煤直接液化工艺中硫元素的回收利用[J].洁净煤技术,2016,22(5):95-99.
[3]常卫科,徐洁,孙伟,等.煤液化残渣中硫的迁移和转化研究现状及展望[J].洁净煤技术,2017,23(3):1-6.
[4]吴琼,高宇龙,刘柯澜.神华煤直接液化工艺中硫元素的转化[J].洁净煤技术,2016,22(6):40-45,51
[5]苗强.煤直接液化残渣萃取技术现状及发展趋势[J].洁净煤技术,2015,21(1):56-60.
[6]LI J,YANG J,LIU Z.Hydro-treatment of a direct coal liquefaction residue and its components[J].Catalysis Today,2008,130(2):389-394.
[7]高鹏,孙任晖,刘爱国,等.神东煤与煤液化残渣共热解研究[J].洁净煤技术,2016,22(4):121-125.
[8]CUI H,YANG J,LIU Z,et al.Effects of remained catalysts and enriched coal minerals on devolatilization of residual chars from coal liquefaction[J].Fuel,2002,81(11/12):1525-1531.
[9]赵鹏,冯雷,刘盖,等.煤液化残渣在道路沥青混凝土中的应用研究[J].筑路机械与施工机械化,2016,33(2):61-64.
[10]ZHAO C,HUANG D,CHEN J,et al.The interaction of cyanide with pyrite,marcasite and pyrrhotite[J].Minerals Engineering,2016,95:131-137.
[11]王增辉,栾和林,轩小朋,等.硫铁化合物氧化历程中还原硫物质的表征和研究[J].矿冶,2009,18(1):96-99.
[12]卢龙,王汝成,薛纪越,等.硫化物矿物的表面反应及其在矿山环境研究中的应用[J].岩石矿物学杂志,2001,20(4):387-394.
[13]BAHRAMI S,ARDEJANI F D.Prediction of pyrite oxidation in a coal washing waste pile using a hybrid method,coupling artificial neural networks and simulated annealing (ANN/SA)[J].Journal of Cleaner Production,2016,137:1129-1137.
[14]卢龙,王汝成,薛纪越,等.黄铁矿氧化速率的实验研究[J].中国科学,2005,35(5):434-440.
[15]TABELIN C B,VEERAWATTANANUN S,ITO M,et al.Pyrite oxidation in the presence of hematite and alumina:I.batch leaching experiments and kinetic modeling calculations[J].Science of the Total Environment,2016,580:687-698.
[16]TU Z,GUO C,ZHANG T,et al.Investigation of intermediate sulfur species during pyrite oxidation in the presence and absence of acidithiobacillus ferrooxidans[J].Hydrometallurgy,2016,167:58-65.
[17]COOMBS P G,MUNIR Z A.The mechanism of oxidation of ferrous sulfide(FeS)powders in the range of 648 to 923 K[J].Metallurgical Transactions B,1989,20(5):661-670.
[18]CHENG Z L,BAO S C,ZHANG S T,et al.Characteristic of thermal decomposition kinetics of main gold-bearing sulfides pyrite[J].Chinese Journal of Nonferrous Metals,2015,25(8):2212-2217.
[19]ZHAO C,CHEN J,LI Y,et al.First-principle calculations of interaction of O2with pyrite,marcasite and pyrrhotite surfaces[J].Transactions of Nonferrous Metals Society of China,2016,26(2):519-526.
[20]FOGLER H S.Elements of chemical reaction engineering[M].4th.Englewood:Prentice-Hall PTR,2006:788-797.
[21]KATIRCIOGLU T Y,KAPTAN H Y,GUVEN O.Determination of oxygen diffusion coefficient of poly(methacrylonitrile)II and the calculation of diffusion activation energy[J].Journal of Applied Polymer Science,2015,74(5):1108-1118.
[22]WU P, ZENG Y, JIN H M.Interpretation and prediction of diffusion activation energy from thermodynamic model[J].Springer Singapore,2015,204(16):149-157.
[23]VERMAAT J E,HARMSEN J,HELLMANN F A,et al.Annual sulfate budgets for dutch lowland peat polders:the soil is a major sulfate source through peat and pyrite oxidation[J].Journal of Hydrology,2015,533:515-522.
[24]WU P,ZEMG Y,JIN H M.Interpretation and prediction of diffusion activation energy from thermodynamic model[M].Berlin:Springer Singapore,2015:690-696.
猜你喜欢
山东冶金(2022年3期)2022-07-19 03:27:06
昆钢科技(2022年2期)2022-07-08 06:36:28
云南化工(2021年9期)2021-12-21 07:43:42
山东冶金(2019年3期)2019-07-10 00:53:54
航海(2018年1期)2018-03-08 20:18:39
江苏理工学院学报(2017年2期)2017-07-09 21:02:05
当代贵州(2017年5期)2017-04-11 11:08:21
咸阳师范学院学报(2016年6期)2017-01-15 14:18:46
塑料包装(2015年2期)2015-12-20 08:08:48
化工进展(2015年3期)2015-11-11 09:09:01
