
硫酸根离子强化零价铁去除Cr(Ⅵ)的试验研究
2018-04-25 13:08胡艺泓黄廷林孙远奎西安建筑科技大学环境与市政工程学院陕西西安70055同济大学环境科学与工程学院上海200092
中国环境科学 2018年4期
胡艺泓,黄廷林,孙远奎 (.西安建筑科技大学环境与市政工程学院,陕西 西安 70055;2.同济大学环境科学与工程学院,上海 200092)
铬是人们普遍关注的致癌物质[1-2],冶金、制革、纺织品生产、印染、颜料以及电镀等行业排放的含铬废水是铬污染的主要来源[3-5].水体中铬通常以无机态的Cr(Ⅵ)和Cr(Ⅲ)形式存在[6-7].其中,Cr(Ⅵ)是一种潜在的致癌物质,有较高的迁移性和很强的毒性[8].通常零价铁(Fe0)在水中发生一系列腐蚀反应生成 Fe2+[9-11],其与 Fe0可将Cr(Ⅵ)还原为毒性较小的 Cr(Ⅲ)[12-14],Cr(Ⅲ)可进一步转化为Cr(OH)3沉淀或与Fe(Ⅲ)形成配合物而从水中去除[15-16].
然而, Fe0在生产过程中表面会形成一层铁氧化物膜使其活性较低[17],且 Cr(Ⅵ)也会导致Fe0较快钝化.因此,提高 Fe0的反应活性对推动Fe0的实际应用具有重要意义.大量研究表明,水中共存阴离子可显著影响 Fe0的反应活性[18-19],其中一些阴离子能够与Fe0表面发生反应而使其钝化[20-22],另一些则会攻击其钝化膜从而促进腐蚀[23].最近研究[24]发现与 Cl-、ClO4-、NO3-等离子相比,SO42-能显著提高 Fe0除 As(Ⅲ)的性能且随着浓度增大而促进作用愈加明显,但当浓度增加到一定程度后会趋于稳定,促进效果不再有明显上升.同样,前人在研究 Fe0去除 4-氯硝基苯的反应活性影响时也发现,SO42-能大大提高去除效果[25].虽然上述结果表明 SO42-也许可以促进零价铁去除 Cr(VI)的效果,但考虑到前人研究多采用间歇实验来考察共存离子的影响,且未全面监测反应过程中其他相关指标变化,不能完全反映共存离子在连续流、长期运行条件下的作用.因此本文拟通过柱子实验来研究连续流条件下硫酸根离子对零价铁柱去除水中 Cr(VI)的作用,分析硫酸根离子存在时零价铁去除Cr(VI)的机理.
1 材料与方法
1.1 实验材料
实验所用铁粉购自深圳市创辉磁材厂,粒径100目,其表面形貌如图 1所示.实验所用试剂主要有:NaCl、Na2SO4、Na2SiO3、NaH2PO4、K2Cr2O7、二苯基碳酰二肼等均为分析纯级别,对应试剂储备液均采用 ELGA超纯水机提供的超纯水配制,4℃下保存.

图1 原始零价铁表面形貌Fig.1 SEM image of the pristine ZVI
1.2 实验方法
实验用水由蒸馏水配制,且加入 3mmol/L NaCl、0.1mmol/L Na2SiO3、0.01mmol/L NaH2PO4以模拟地下水.反应开始时不加入 SO42-,后续以Na2SO4的形式加入,为不超出饮用水标准的限制,硫酸根离子的浓度控制在 1~3mmol/L.进水初始pH 值为 9.0~9.5,DO 约为 5.8mg/L,初始 Cr(Ⅵ)浓度为1mg/L.
实验装置为长300mm、内径为16mm的有机玻璃柱,柱子上下两端分别填充50mm的粒径为6~10目的粗砂,中间200mm填充铁粉(30g)和细砂(45g)的混合物,细沙粒径为 40目.上向流进水,流速为 3mL/min.反应启动后,按照一定的时间间隔收集出水,用注射器取10mL过0.22 µm滤头,加酸酸化后保存待测.总铬及总溶解性铁使用ICP-MS测定,Cr(Ⅵ)使用二苯基碳酰二肼分光光度计法测定.所收集的出水同时测定pH值、DO值.pH值使用奥豪斯 STARTER 2100pH 计测定,DO测定使用雷磁JPBJ-608溶解氧仪.反应结束后,分别收集柱子上、中、下部的固相物质,并利用场发射扫描电子显微镜(SEM)和 X射线光电子能谱仪(XPS)进行表征,XPS结果采用AvantageXPS软件分析.
2 结果与讨论
2.1 硫酸根离子对零价铁柱去除水中 Cr(Ⅵ)的影响
硫酸根投加前后零价铁柱去除水中 Cr(Ⅵ)的运行效果如图2所示.可以看到,在运行初期的5.5h内,柱子出水中基本未检测到 Cr的存在,说明此时 Fe0可有效去除水中的 Cr(VI).然而随着反应的进行,出水中的Cr(VI)及总Cr的浓度逐渐升高并超出了饮用水的最高允许浓度(50 µg/L).需要指出的是,在这一过程中出水中的总Cr浓度要高于出水中 Cr(VI)的浓度,这说明出水中含有其他价态的溶解性Cr,结合前人的研究结果推测其应为Cr(III),这也可通过后续的XPS表征得到验证.随着时间的推移,出水中的 Cr逐渐增加,在运行 49.5h后,出水中的总 Cr浓度已高达~630µg/L,而此时柱子中仍含有大量未反应的Fe0(总投量为 30g).即,随着反应的进行 Fe0的反应活性逐渐降低,其已不能有效地去除水中的Cr(VI),这造成了 Fe0的大量浪费.Hu 等人[26]也曾报道Fe0还原Cr(Ⅵ)的过程是一个自抑制的过程,这是由于反应过程中生成的铁氢氧化物及铬氢氧化物膜会覆盖在Fe0表面阻碍Cr(Ⅵ)与零价铁的进一步接触.
为了解决这一问题,本研究尝试向水中投加SO42-来提高Fe0去除Cr(VI)的反应活性.具体来说,在运行 49.5h后,向进水中投加 1mmol/L的SO42-(为保证处理效果,在286h时将SO42-的浓度提高至约3mmol/L)并监测水中Cr(VI)、总Cr等参数的变化.如图2所示,在加入SO42-后,出水中的Cr(VI)和总Cr开始逐渐降低:投加SO42-仅3h后,出水中的总Cr即已降低至268µg/L;而经过约50h后,出水中的Cr(VI)、总Cr进一步降低至《生活饮用水卫生标准》(GB5749-2006)[27]所规定的限值50µg/L以下,此后的240h内这一处理效果保持稳定.这些说明硫酸根离子确实能提高已钝化Fe0的反应活性并能缓解Cr(VI)对Fe0的致钝效应.

图2 出水中总Cr和Cr(VI)随运行时间的变化规律Fig.2 The variation of residual total Cr and Cr(VI) in effluent with running time
2.2 反应过程pH值、DO及总溶解性Fe的变化规律
在 Fe0体系中加入硫酸根离子后,Cr(VI)去除效果的提高说明Fe0的腐蚀得到了加速.为了更好地了解Fe0柱去除Cr(VI)的反应过程,本研究还监测了反应过程中溶液pH值、DO值及总溶解性Fe的变化规律,结果如图 3所示.在有氧的条件下,Fe0被水中的溶解氧氧化生成 Fe2+[式(1)],同时过程中还将伴随着pH值的上升.随着反应的进行,溶液中的Fe2+会继续被氧气氧化生成Fe3+[式(2)],紧接着Fe3+发生水解生成Fe(OH)3沉淀[式(3)],而Fe3+水解过程中将伴随着pH值的降低.如图3a所示,与进水pH值(9.0~9.5)相比,不加硫酸根离子时出水pH值略有上升而加入硫酸根离子后出水pH值没有明显的变化,这主要是由于在未投加硫酸根离子前,零价铁腐蚀[式(1)]、Fe2+氧化速度[式(2)]快于Fe3+水解[式(3)]速度所致.
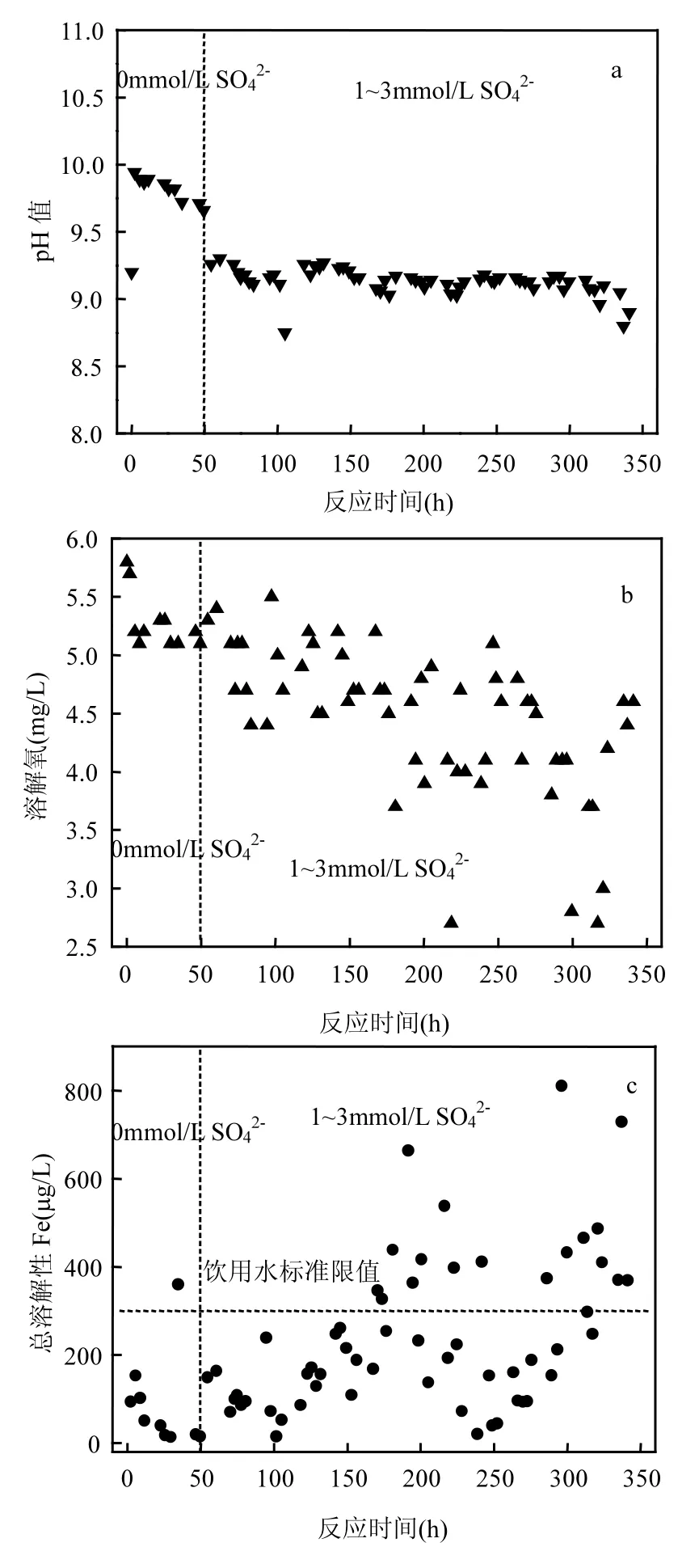
图3 反应过程中各指标随运行时间的变化规律Fig.3 The variation of (a) pH, (b) DO, (c) total dissolved Fe during the running time

如图 3b所示,因发生有氧腐蚀,出水中的DO总体呈降低趋势:与原水DO相比,不加硫酸根离子时出水DO从5.8mg/L约降低至5.0mg/L;而在加入硫酸根离子后,出水DO的波动更为明显且其最高可降低至2.5mg/L.这一区别进一步说明硫酸根离子的加入能促进 Fe0的腐蚀进而可提高其去除 Cr(VI)的反应活性.Fe0的腐蚀将向溶液中释放 Fe2+,尽管碱性条件下,Fe2+的能迅速被氧化成 Fe3+并形成沉淀,但溶液中仍难免会残留部分溶解性铁.如图3c所示,本实验中的出水溶解性铁浓度相对较低(0~800µg/L),在加入硫酸根离子后,溶解性总铁的浓度会有所升高并有可能超出饮用水限值.但考虑到 Fe2+的去除比较容易,在实际应用过程中其可通过曝气或加大柱子上方石英砂厚度的方式得以解决.
2.3 Fe0腐蚀固相产物的形貌分析


图4 反应结束后固相产物SEM图Fig.4 SEM images of solid corrosion products collected at (a) Top layer, (b) Middle layer, (c) Bottom layer
为更好地了解反应前后Fe0表面形貌的变化,在反应结束后分别收集了上、中、下不同位置的固相物质并进行了SEM分析.由图 4可以看到,与原始零价铁相比,反应后的 Fe0表面有球状或粒状的腐蚀产物生成,而且沿水流方向自下而上腐蚀产物的量越来越少,这说明下部零价铁的腐蚀程度更高.此外,仔细观察各部分Fe0的SEM图还可发现, Fe0表面有网状及片状腐蚀产物生成,其中在图4b中还观察到了一个较完美六边形铁氧化物,结合这一独特形状及本文的实验条件可推测其应为硫酸盐型绿铁锈[Fe4IIFe2III(OH)12SO4⋅yH2O],其相关生成特性及对除铬效果的影响有待进一步研究.
2.4 硫酸根存在时Fe0去除Cr(VI)的机理
目前,国内外研究者普遍认为 Fe0除铬的机理为:Fe0在水中发生电化学腐蚀产生Fe2+,Cr(Ⅵ)被 Fe0和Fe2+还原为 Cr(Ⅲ),生成的 Cr(Ⅲ)可在水中形成微溶的Cr(OH)3或Fe-Cr配合物而从水中分离.主要相关反应方程式如下:
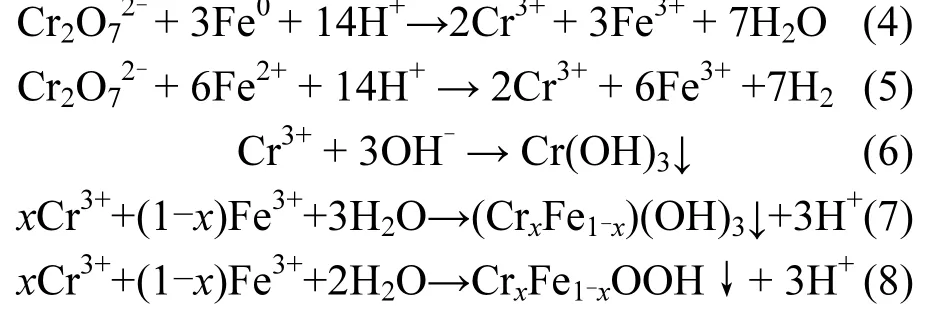
因此,为了探明硫酸根存在时Fe0去除Cr(VI)的机理,本文对反应结束后的固相物质进行了XPS分析,结果如图5所示.由图5b和图5c可以看到,中部和下部Fe0的XPS谱图中光电子峰均在576.9eV(Cr 2p3/2)及 586.7eV(Cr 2p1/2)处出现,而此结合能及谱线与 Cr(Ⅲ)的结合能及线型对应.此外,两个峰之间的自旋轨道分裂值为9.8eV,半峰宽为3.0eV,这些都是Cr(Ⅲ)的特征.因此可以推断本实验固体样品中的 Cr主要是以 Cr(Ⅲ)的形式存在,即 Cr(Ⅵ)可被 Fe0柱还原为 Cr(Ⅲ)而被去除.另一方面,尽管 SO42-能够强化零价铁的反应活性,但其并未改变零价铁去除Cr(VI)的机理.
通过对比图5中Cr(Ⅲ)的峰强度及峰面积还可以发现,下部样品中 Cr(Ⅲ)的峰强度最高,中部样品次之而上部样品基本没有Cr(III)的峰.这一现象说明在本实验的运行时间内,Cr(VI)的去除主要是通过中下部的 Fe0来实现的,而上部的 Fe0还未得到充分利用.这也与图 2中在 Fe0填充柱运行近350h后Cr(VI)依然能被有效去除相一致.


图5 反应结束后固相产物的Cr 2p XPS谱图Fig.5 XPS survey of chromium 2p of solid corrosion products after reaction. (a) Top layer, (b) Middle layer, (c) Bottom layer
2.5 硫酸根提高零价铁反应活性的机理探讨
有研究表明[28],硫酸根离子能促进零价铁的腐蚀,进而能提高 Fe0去除某些污染物的反应活性.本文的实验结果也证明,在硫酸根离子加入后,Fe0的腐蚀得到了加速(图 3).但到目前为止,关于硫酸根离子如何加速 Fe0腐蚀的机理尚不清晰.其中,较可能的解释之一是SO42-能促进Fe0表面钝化膜的溶解,其反应式如公式9.具体来说,硫酸根离子可作为配体(L-)取代铁氧化物表面的羟基并与表面的铁形成溶解态的单齿或双齿配合物,这一过程导致钝化膜的破坏从而能维持或加速Fe0铁的腐蚀[29-30].

3 结论
3.1 当原水中没有硫酸根离子存在时,Fe0柱去除Cr(Ⅵ)的活性较差,运行5.5h后出水总铬浓度即已超出饮用水标准限值(50µg/L);向原水中投加 1~3mmol/L的硫酸根离子后出水总铬浓度逐渐降低至50µg/L以下,并保持这一效果长达240h以上.由此可见,硫酸根离子能大大提高零价铁去除 Cr(Ⅵ)的反应活性,进而使 Fe0柱的出水铬浓度能满足国家饮用水标准,且该效果具有长效稳定性.
3.2 向 Fe0体系中加入硫酸根离子后,溶解氧的消耗(最高可从 9.5mg/L降低至2.5mg/L)及铁氧化物的生成均得到了提高,表明硫酸根离子能加速Fe0的腐蚀.
3.3 XPS的分析结果显示固相产物中的 Cr主要以Cr(III)形式存在,说明Fe0去除Cr(Ⅵ)的机理主要为还原作用,且硫酸根离子的加入并未改变此机理,尽管其能够强化Fe0活性.
参考文献:
[1]EPA. U. S. Field applications of in situ remediation technologies:Permeable reactive barriers [M]. Washing, DC: EPA, 1999.
[2]Katz S, Salem H. The biological and environmental chemistry of chromium [M]. Weinheim: VCH Publisher, 1994.
[3]Auden W H. Handbook of groundwater remediation using permeable reactive barriers-applications to radionuclides, trace metals, and nutrients [M]. 2002.
[4]Pratt A, Blowes D, Ptacek C. Products of chromate reduction on proposed subsurface remediation material [J]. Environmental Science & Technology, 1997,31(31):2492-2498.
[5]周 密.腐殖酸对零价铁去除污染水体中六价铬的影响 [D].杭州:浙江大学, 2007.
[6]秦泽敏,董黎明,刘 平,等.零价纳米铁吸附去除水中铬的研究[J]. 中国环境科学, 2014,34(12):3106-3111.
[7]张 珍,沈珍妙,范天恩,等.腐殖酸对nZVI去除水中Cr(Ⅵ)的抑制及抑制作用的消除 [J]. 中国环境科学, 2013,33(1):63-68.
[8]Zazo J, Paull J, Jaffe P. Influence of plants on the reduction of hexavalent chromium in wetland sediments [J]. Environmental Pollution, 2008,156(1):29-35.
[9]Guan X, Sun Y, Qin H, et al. The limitations of applying zero-valent iron technology in contaminants sequestration and the corresponding countermeasures: The development in zero-valent iron technology in the last two decades (1994~2014) [J]. Water Research, 2015,75:224-248.
[10]Noubactep C. The fundamental mechanism of aqueous contaminant removal by metallic iron [J]. Water Sa, 2010,4738(4738):663-670.
[11]孙远奎.水中 As(III)的短程去除新方法及其反应机制研究 [D].上海:同济大学, 2015.
[12]杜珮雯,党宏钰,张永祥,等.改性沸石和铁粉的复合材料理水中六价铬的研究 [J]. 中国环境科学, 2015,35(5):1384-1390.
[13]聂 宁,丁远昭,李喜青.制备斜发沸石和零价铁复合材料处水中的六价铬污染 [J]. 中国环境科学, 2013,33(3):443-447.
[14]Guertin J, Jacobs J, Avakian C, et al. Chromium (vi) handbook[M]. Florida: CRC Press, 2004.
[15]Li X, Cao J, Zhang W. Stoichiometry of Cr(VI) immobilization using nanoscale zerovalent Iron (nZVI): A study with high-resolution X-ray photoelectron spectroscopy (HR-XPS) [J]. Industrial & Engineering Chemistry Research, 2008,47(7):2131-2139.
[16]王 吟,赵建夫,王学江,等.零价铁修复铬污染水体研究进展[J]. 安徽农业科学, 2010,38(6):3117-3119.
[17]Gheju M. Hexavalent chromium reduction with zero-valent iron(ZVI) in aquatic systems [J]. Water Air & Soil Pollution,2011,222(1-4):103-148.
[18]Farrell J, Kason M, Melitas N, et al. Investigation of the long-term Performance of zero-valent iron for reductive dechlorination of trichloroethylene [J]. Environmental Science &Technology, 2000,34(3):514-521.
[19]Klausen J, Ranke J, Schwarzenbach R. Influence of solution composition and column aging on the reduction of nitroaromatic compounds by zero-valent iron [J]. Chemosphere, 2001,44(4):511-517.
[20]Kohn T, Roberts A L. The effect of silica on the degradation of organohalides in granular iron columns [J]. Journal of Contaminant Hydrology, 2006,83(1/2):70-88.
[21]Kohn T, Kane S, Fairbrother D, et al. Investigation of the inhibitory effect of silica on the degradation of 1,1,1-trichloroethane by granular iron [J]. Environmental Science &Technology, 2003,37(24):5806-5812.
[22]Su C, Puls R. Arsenate and arsenite removal by zerovalent iron:effects of phosphate, silicate, carbonate, borate, sulfate, chromate,molybdate, and nitrate, relative to chloride [J]. Environmental Science & Technology, 2001,35(22):4562-4568.
[23]Gotpagar J, Lyuksyutov S, Cohn R, et al. Reductive dehalogenation of trichloroethylene with zero-valent iron: surface profiling microscopy and rate enhancement studies [J]. Langmuir,1999,15(24):8412-8420.
[24]Sun Y, Hu Y, Huang T, et al. Combined effect of weak magnetic fields and anions on arsenite sequestration by zerovalent Iron:kinetics and mechanisms [J]. Environmental Science &Technology, 2017,51(7):3742-3750.
[25]Devlin J, Allin K. Major anion effects on the kinetics and reactivity of granular iron in glass-encased magnet batch reactor experiments [J]. Environmental Science & Technology, 2005,39(6):1868-1874.
[26]Hu C, Lo S, Liou Y, et al. Hexavalent chromium removal from near natural water by copper-iron bimetallic particles [J]. Water Research, 2010,44(10):3101-3108.
[27]GB5749-2006 生活饮用水卫生标准 [S].
[28]Sun Y, Li J, Huang T, et al. The influences of iron characteristics,operating conditions and solution chemistry on contaminants removal by zero-valent iron: A review [J]. Water Research, 2016,100:277-295.
[29]Zhu M, Northrup P, Shi C, et al. Structure of sulfate adsorption complexes on ferrihydrite [J]. Environmental Science &Technology Letters, 2014,1(1):97-101.
[30]Jeong D, Kim K, Min D, et al. Freezing enhanced dissolution of iron oxides: Effects of inorganic acid anions [J]. Environmental Science & Technology, 2015,49(21):12816-12822.
猜你喜欢
黑龙江水利科技(2022年9期)2022-10-13
土壤学报(2022年3期)2022-08-26
环境工程技术学报(2022年3期)2022-06-05
建材发展导向(2021年14期)2021-08-23
世界有色金属(2021年6期)2021-06-14
环境卫生工程(2020年3期)2020-07-27
江苏科技报·E教中国(2020年4期)2020-07-08
中国煤层气(2019年2期)2019-08-27
中国水利(2015年21期)2015-01-30
火炸药学报(2014年1期)2014-03-20
