
保护品种云茶1号茶树全长转录组测序分析
2018-04-18 05:20朱兴正夏丽飞陈林波孙云南田易萍宋维希蒋会兵
茶叶科学 2018年2期
朱兴正,夏丽飞,陈林波*,孙云南,田易萍,宋维希,蒋会兵
保护品种云茶1号茶树全长转录组测序分析
朱兴正1,2,夏丽飞1,2,陈林波1,2*,孙云南1,2,田易萍1,2,宋维希1,2,蒋会兵1,2
1. 云南省农业科学院茶叶研究所/云南省茶树种质资源创新与配套栽培技术工程研究中心,云南 勐海 666201; 2. 云南省茶学重点实验室,云南 勐海 666201
为探讨云茶1号茶树品种优异性状的遗传基础,采用PacBio平台进行全长转录组测序,最终获得Polished consensus序列213 389个,预测到CDS有223 120个,检测到195 062个SSR位点。在NR数据库有170 264个同源序列比对到980个物种;有103 124个在KOG数据库得到注释,根据其功能各分为26类;有65 524个得到GO注释分为细胞组分、分子功能及生物学过程等三大类的55个功能组;根据KEGG数据库,105 972个得到了注释,涉及到216个代谢途径分支,包括茶叶品质、活性物质代谢以及抗逆等相关基因等;还预测到隶属于60个转录因子家族的转录因子有5 785个。这些结果为进一步开展云茶1号茶树特异性状基因的标记性引物开发、遗传研究以及品质形成和抗逆机制研究奠定了基础。
云茶1号;全长转录组;基因分析;功能注释
云茶1号茶树品种是云南省农业科学院茶叶研究所于1993—2005年从云南元江细叶糯茶群体品种中采用单株育种法育成,其芽叶生育力强、持嫩性好,发芽密,茸毛特多,芽叶肥壮,一芽二叶百芽重达144.0 g。春茶一芽二叶蒸青样水浸出物44.6%,茶多酚31.3%,氨基酸3.4%,咖啡碱4.3%,儿茶素总量158.7 mg·g-1,适制红茶、绿茶、普洱茶、白茶。抗逆性鉴定发现其抗寒、抗旱、抗小绿叶蝉和抗茶饼病能力强,在云南的西双版纳、普洱、保山等州(市)有种植,湖南、广西等省有引种,是一个性状优异的优良品种[1-3]。2005年11月国家林业局植物新品种保护办公室授予植物新品种权,品种权号20050030[4]。
基于PacBio平台的第三代测序技术是一种集高通量、快速度以及超长的读长优势和低成本等多种优点于一身的新型测序技术。它最大特点是无需进行PCR扩增,可直接读取目标序列,因此假阳性率大大减少,同时避免了碱基替换及偏置等常见PCR错误的发生,精准度可达到99.9%[5-6]。因此,本试验采用PacBio平台的第三代测序技术对云茶1号进行全长转录组测序,通过生物信息学方法对所测的序列进行序列分析、功能注释、功能分类及代谢途径分析等,旨在为进一步挖掘云茶1号茶树品种次生代谢相关功能基因、抗逆基因及开发分子标记奠定基础。
1 材料与方法
1.1 试验材料
采摘云南省农业科学院茶叶研究所试验基地树龄为13年的云茶1号茶树的芽、叶、茎、花芽、花蕾、幼果,利用液氮迅速固样后送至北京诺禾致源科技股份有限公司进行RNA提取及测序等分析。
1.2 RNA的提取与文库构建
采用Trizol法[7]提取分别提取云茶1号芽、叶、茎、花芽、花蕾、幼果总RNA,利用琼脂糖凝胶电泳、Nanodrop、Agilent 2100、Qubit等检测合格后,进行等量混匀。混匀后的RNA利用带有Oligo(dT)的磁珠富集mRNA,使用SMARTer PCR cDNA Synthesis Kit将mRNA反转录为cDNA,再用BluePippin进行片段筛选,对全长cDNA进行损伤修复、末端修复、连接SMRT哑铃型接头和核酸外切酶消化后获得文库。
1.3全长转录组测序及序列分析
对库检合格文库运用Pacbio Sequel平台进行测序,原始下机数据采用PacBio官方软件SMRTlink处理获得Subreads序列,由Subreads之间的校正获得环形一致性序列(CCS),再根据序列是否包含5'端引物、3'端引物以及polyA尾将序列分为全长序列与非全长序列,然后利用同种类型聚类(ICE)对全长序列进行聚类,获得Cluster consensus序列,最后使用非全长序列对得到的一致序列进行校正(Polishing),获得高质量的序列进行后续分析。再利用ANGEL软件对获得的序列进行编码蛋白产物的序列(Coding sequence, CDS)预测分析[8]。采用MISA软件对单核苷酸、二核苷酸、三核苷酸、四核苷酸、五核苷酸和六核苷酸按照最少重复次数分别为9、6、5、5、5和5对Gene进行SSR检测。
1.4全长转录组序列的功能注释
对获得的高质量序列,利用公共数据库包括非冗余蛋白数据库(Non-Redundant Protein Database, NR)、蛋白质真核同源数据库(Eukaryotic Orthologous Groups, KOG)、蛋白质序列数据库(Swiss Prot Protein Database, Swiss Prot)、蛋白质原核同源数据库(Cluster of Orthologous Groups of Proteins, COG)、基因本体论数据库(Gene Ontology, GO)、蛋白质家族域数据库(Protein Families Database, Pfam)、东京基因与基金组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG)进行基因功能注释。使用iTAK软件[9]以及采用database中分类定义好的转录因子(Transcription Factor, TF)family及规则,通过hmmscan鉴定TF。
2 结果与分析
2.1 全长转录组测序与组装分析
2.1.1 测序结果与数据组装
采用PacBio Sequel测序平台对云茶1号芽、叶、茎、花芽、花蕾、果等混合样进行全长转录组测序,共获得6 282 894个Subreads(10.62 Gb),平均subreads长度为1 690 bp,N50为2 646。通过检测其中环形一致性序列(Circular Consensus Sequence, CCS)Read有499 432个,带有5'端引物的reads数为454 699个,带有3'端引物的reads数有464 035个,带有PolyA尾的reads数有451 234个;全长(Full-Length, FL)reads数有400 959个;全长非嵌合(Full-Length non-chimericRead, FLNC)reads数有388 370,FLNC平均长度为2 469 bp;使用ICE算法将同一转录本的FLNC序列进行聚类,得到consensus序列,并采用非全长的序列对得到的consensus序列进行校正,最终获得Polished consensus序列213 389个,用于后续分析。
2.1.2 CDS预测
CDS(Coding sequence)是编码一段蛋白产物的序列,与蛋白质的密码子完全对应。利用ANGEL软件[8]进行CDS预测分析,得到其编码区的氨基酸序列和核酸序列。将获得的Polished consensus在蛋白质数据库中进行Blast比对以后,发现有223 120个Coding Sequence(CDS),序列长度范围分布在200~ 2 500 bp之间,主要集中在300~1 100 bp(图1),说明Unigenes的序列质量较好。
2.1.3 SSR分析
采用MISA对云茶1号全长转录组Gene进行SSR检测,搜索标准为:1~6个核苷酸基序(motif)重复次数分别为大于等于9、6、5、5、5和5,结果见表1。有195 062个序列检测到SSR位点,单核苷酸和二核苷酸重复类型占优势,分别占总SSR的57.58%和31.14%;其他4种重复类型所占比例相对较少:三核苷酸重复占9.28%,四核苷酸重复占0.92%,五核苷酸重复占0.46%,六核苷酸重复占0.62%。在检测到的SSR位点中,单核苷酸中出现频率最高的核苷酸基序为T/A(94 710)和A/T(45 750个);二核苷酸以CT/AG最多有13 993个,其次是TC/GA和AG/CT,分别有13 492个和8 246个;三核苷酸以GAA/CTT(956个)、TTA/AAT(919个)、CCA/GGT(875个)占优势;四核苷酸出现频率以TTTA/AAAT(296个)和TTAT/AATA(133个)为多;五核苷酸和六核苷酸分别以TGTGT/ACACA(159个)和TCCACC/ AGGTGG(25个)最多。云茶1号中儿茶素、氨基酸、咖啡碱等茶叶品质成分含量高、抗逆性强以及芽叶大等优良性状,研究价值很高。因此,上述对SSR特征分析的结果,为进一步开展云茶1号及茶树通用性引物设计及其遗传图谱构建等打下了良好基础。
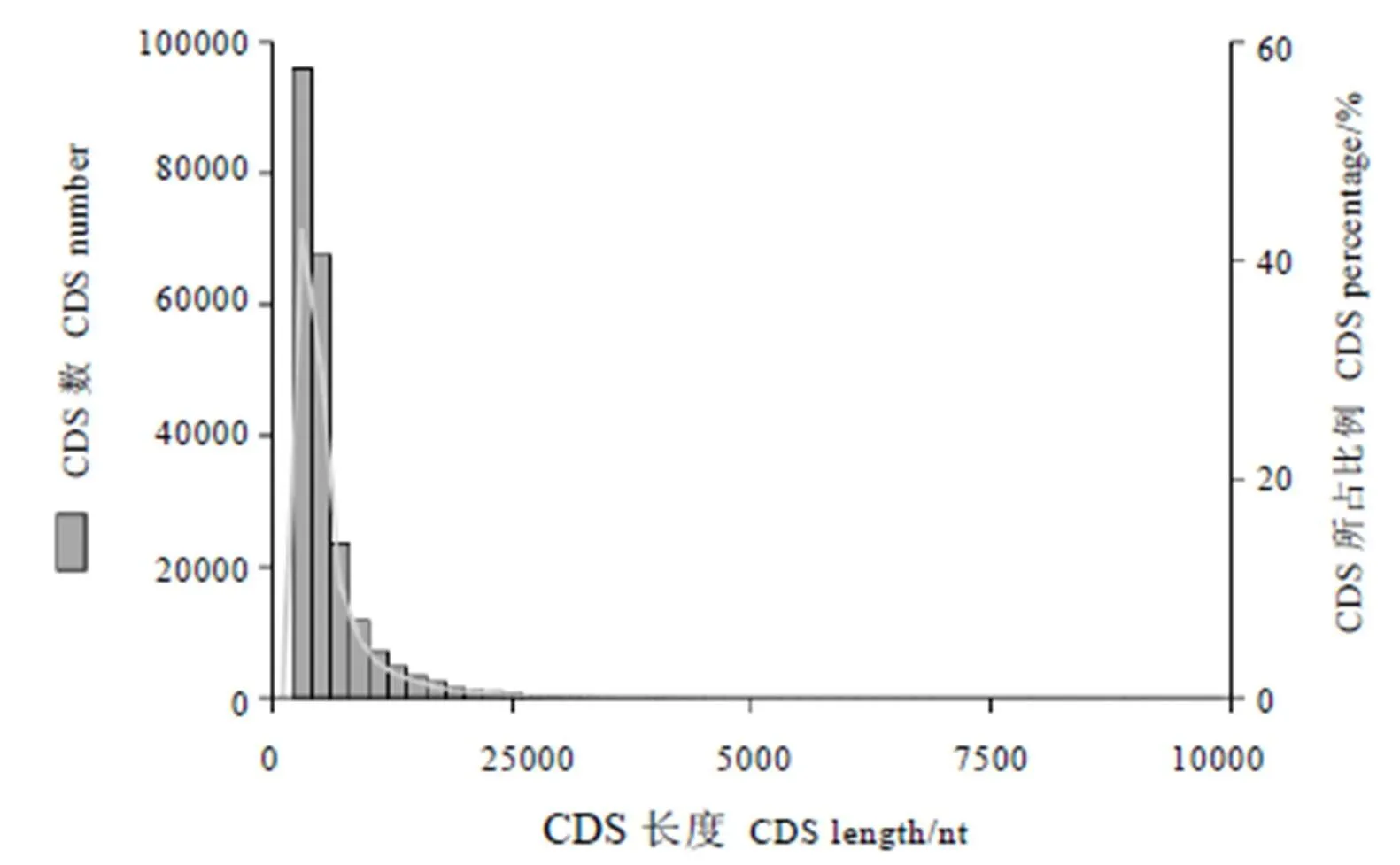
图1 CDS长度分布
Fig.1CDS length distribution
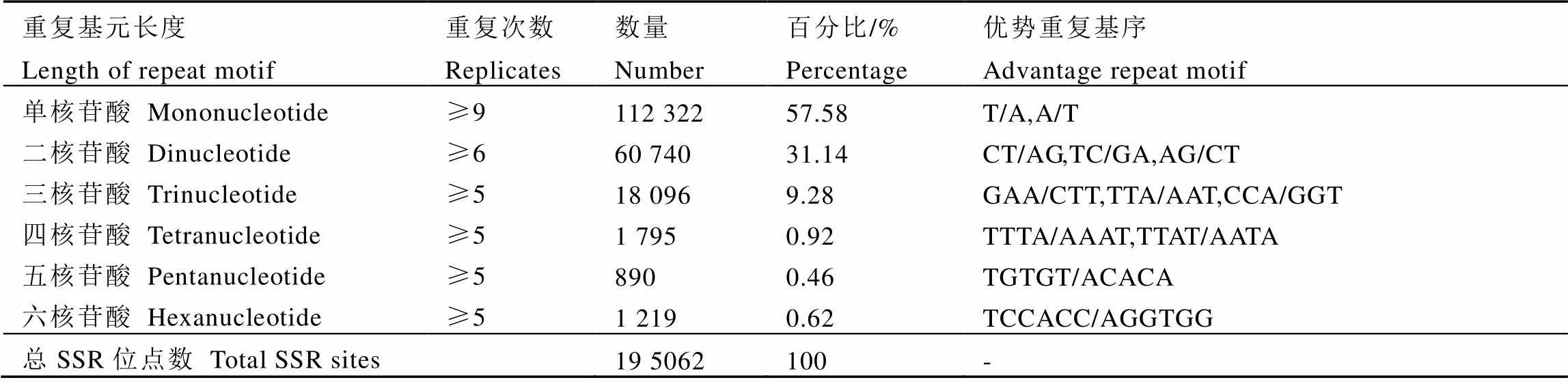
表1 SSR不同重复基序分布及优势碱基组成
2.2 Unigene的功能注释
2.2.1注释结果统计
通过BLAST程序,对转录组序列进行NR数据库比对。有170 264个Unigene在NR数据库比对到980个物种上,比对较多的20个物种包括葡萄(28 588个)、茶树(8 753个)、核桃(7 601个)、中粒咖啡(6 000个)、芝麻(5 892个)、可可(5 767个)、莲(5 471个)、枣(4 170个)、木薯(4 043个)、胡萝卜(3 962个)、麻疯树J(3 499个)、土瓶草(3 335个)、柑橘(3 156个)、牵牛花(3 144个)、蓖麻(3 123个)、桃(2 899个)、野生烟草(2 855个)、杨树(2 663个)、烟草(2 470个)、刺菜蓟(2 330个)、青梅(2 243个)。从NR库中的注释情况看,注释到葡萄的基因最多,可能是GenBank中登录葡萄的基因信息多,且茶树与葡萄的相对较为近缘。同时随着茶树生物学研究的不断深入,登录到GenBank的基因越来越多,获得较多茶树基因注释。
2.2.2 Unigene的KOG分类统计
将云茶1号Unigene与KOG数据库进行比对并根据其功能进行分类统计见表2。有103 124个Unigene得到注释,按照功能一共分为26类,其中一般功能预测最多(19 620个),其次是翻译后修饰、蛋白折叠和分子伴侣(13 209个),最少的为未知蛋白(2个)。在KOG注释分类中,注释到次生代谢物合成、运输和代谢有5 183个,氨基酸运输和代谢的有4 363个,说明了云茶1号茶树中次生代谢和氨基酸代谢较为活跃,这为云茶1号茶树茶叶品质的形成奠定了基础。
2.2.3 Unigene的GO功能注释
有65 524个Unigene注释到GO数据库,可分为细胞组分、分子功能、及生物学过程等3个大类的55个功能组(表3)。其中生物学过程包括25个功能组,以代谢过程(33 845个)、细胞过程(33 284个)、单生物过程(24 567个)、定位(9 572个)的最多,细胞聚集功能最少(1个),其次是节律过程(5个);细胞组分包括20个功能组,以细胞部分(15 349个)、细胞(15 349个)、细胞器(9 900个)、膜结构(8 973个)、膜部分(8 487个)较多,以细胞连接、突触以及突触部分最少均为14个;分子功能包括10个功能组,以结合活性(39 544个)、催化活性(32 858个)最多,金属伴侣蛋白活性为最少(3个)等。由于同个基因可能有多个注释,因此,注释到GO数据库的基因总数大于实际注释到的基因数。

表2 Unigene的KOG系统分类
2.2.4 Unigene的KEGG功能注释
KEGG数据库已建立了一套完整KO注释的系统,整合了基因组、化学分子和生化系统等方面的数据,包括代谢通路(KEGG PATHWAY)、功能模型(KEGG MODULE)、基因序列(KEGG GENES)及基因组(KEGG GENOME)等,可完成新测序物种的基因组或转录组的功能注释。云茶1号茶树转录组有105 972个得到了注释,根据参与的KEGG代谢通路分为三个层次,第一层次分为四大类,第二层为23小类(表4),第三层次为216个代谢途径分支。涉及茶叶滋味相关代谢途径的有氨基酸代谢(8 479个)、黄酮类化合物的生物合成(464个)、黄酮和黄酮醇的生物合成(86个)、咖啡因的代谢(41个)。涉及与香气相关的单萜生物合成(80个)、倍半萜生物合成(168个)、萜类化合物骨架生物合成(378个)。涉及植物激素信号转导(1 772个)、MAPK信号通路(1 194个)、AMPK信号通路(1 149个)等。这些Unigene为今后深入开展云茶1号茶滋味、香气以及抗逆等相关的功能基因研究奠定了基础。

表3 Unigene的GO分类
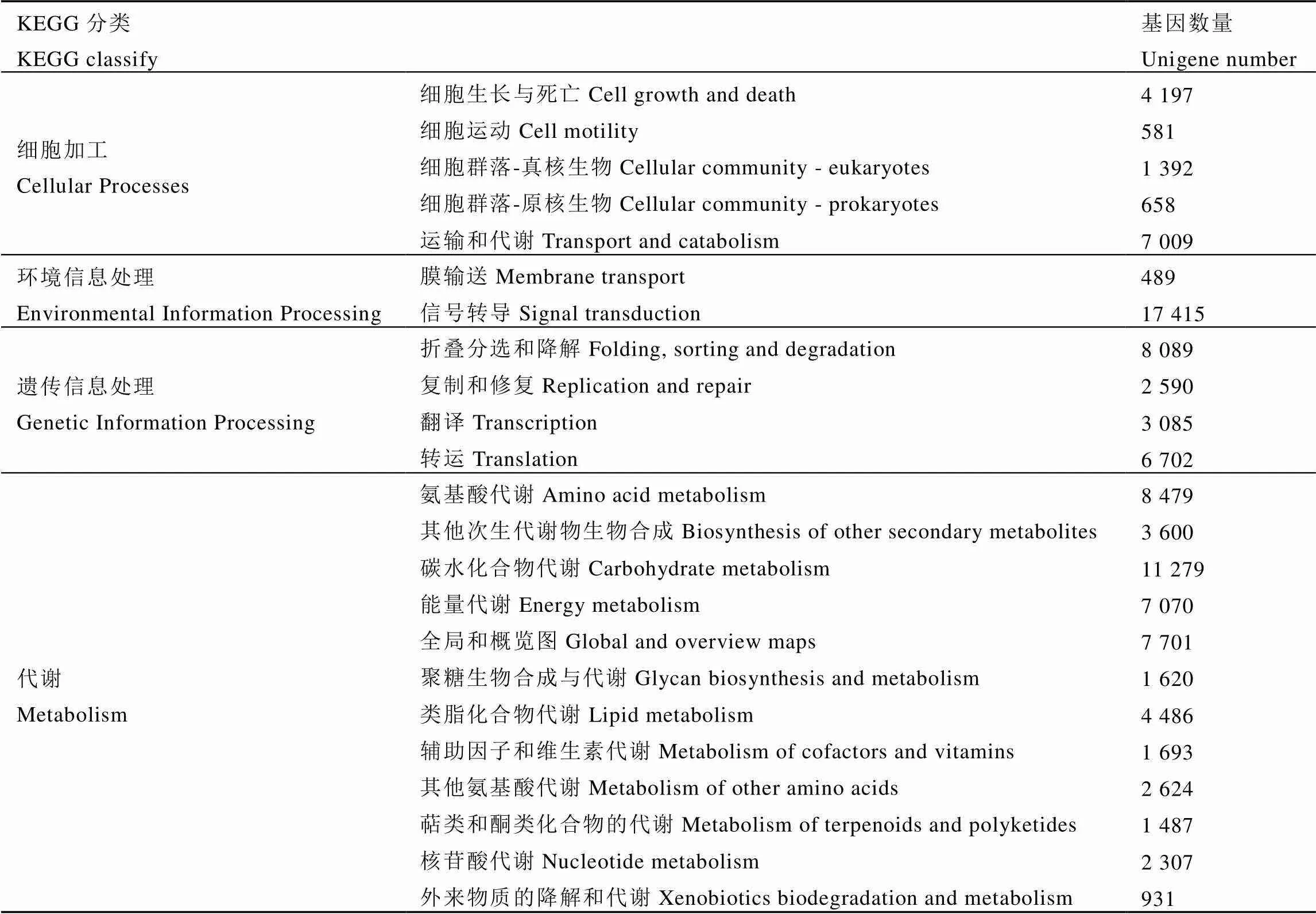
表 4 Unigene的KEGG分类
2.2.5 转录因子分析
转录因子(Transcription factor, TF)也称为反式作用因子,是一群能与基因5'端上游特定序列专一性结合,从而保证目的基因以特定的强度在特定的时间与空间表达的DNA结合蛋白,通过它们之间以及与其他相关蛋白之间的相互作用,激活或抑制转录,在植物生长发育过程、组织分化、营养运输、代谢合成及环境应答等生命过程中起着至关重要的转录调控作用[10]。使用iTAK软件预测到转录因子有5 785个,隶属于60个转录因子家族,其中比较多的转录因子家族见(图2)。在获得的转录因子家族中NAC家族有202个、AP2/ERF家族有185个、AUX/IAA家族有216个、MYB家族有92个、bHLH家族有235个、WRKY家族有172个等。这些转录因子家族成员的获得,为后期研究参与云茶1号茶树中次生代谢物的生物合成、生长发育以及抗逆调控奠定基础。
3 讨论
PacBio第三代测序技术是基于单分子实时(Single molecule real time, SMRT)的单分子测序仪,由美国Pacific Biosciences(PacBio)[5]公司设计制造,最大特点是无需进行PCR扩增,可直接读取目标序列,因此假阳性率大大减少,是转录组从头测序的首选[11-12]。本研究采用PacBio平台对云茶1号茶树全长转录组测序及分析,获得6 282 894个Subreads,平均subreads长度为1 690 bp,N50为2 646,利用公共数据库NR、KOG、Swiss Prot、COG、GO、Pfam、KEGG对获得的213 389个Polished consensus序列进行功能注释和分类。有170 264个Unigene比对到980个物种上;有103 124个Unigene得到KOG注释,按照功能一共分为26类;有105 972个Unigene注释216个代谢途径分支。Shi等[13]采用二代测序技术对龙井43茶树的嫩叶、成熟叶、茎、幼根、花蕾以及成熟种子的总RNA等量混合进行转录组测序,共获得127 094条Unigene,平均长度为355 bp,N50为506 bp,注释到Nr、COG以及KEGG数据库的分别为53 937个、15 701个和16 939个。陈林波等[14]以紫娟茶树的芽、第二叶、开面叶、成熟叶为材料,利用二代测序技术对其转录组进行测序分析,获得206 210条Unigene,注释到NR的为71 799个、注释到GO的为61 141个、注释到KEGG的为30 995个。这些研究结果表明,不论是从获得的序列质量、基因数以及注释到的基因信息来看,第三代测序结果均优于第二代。
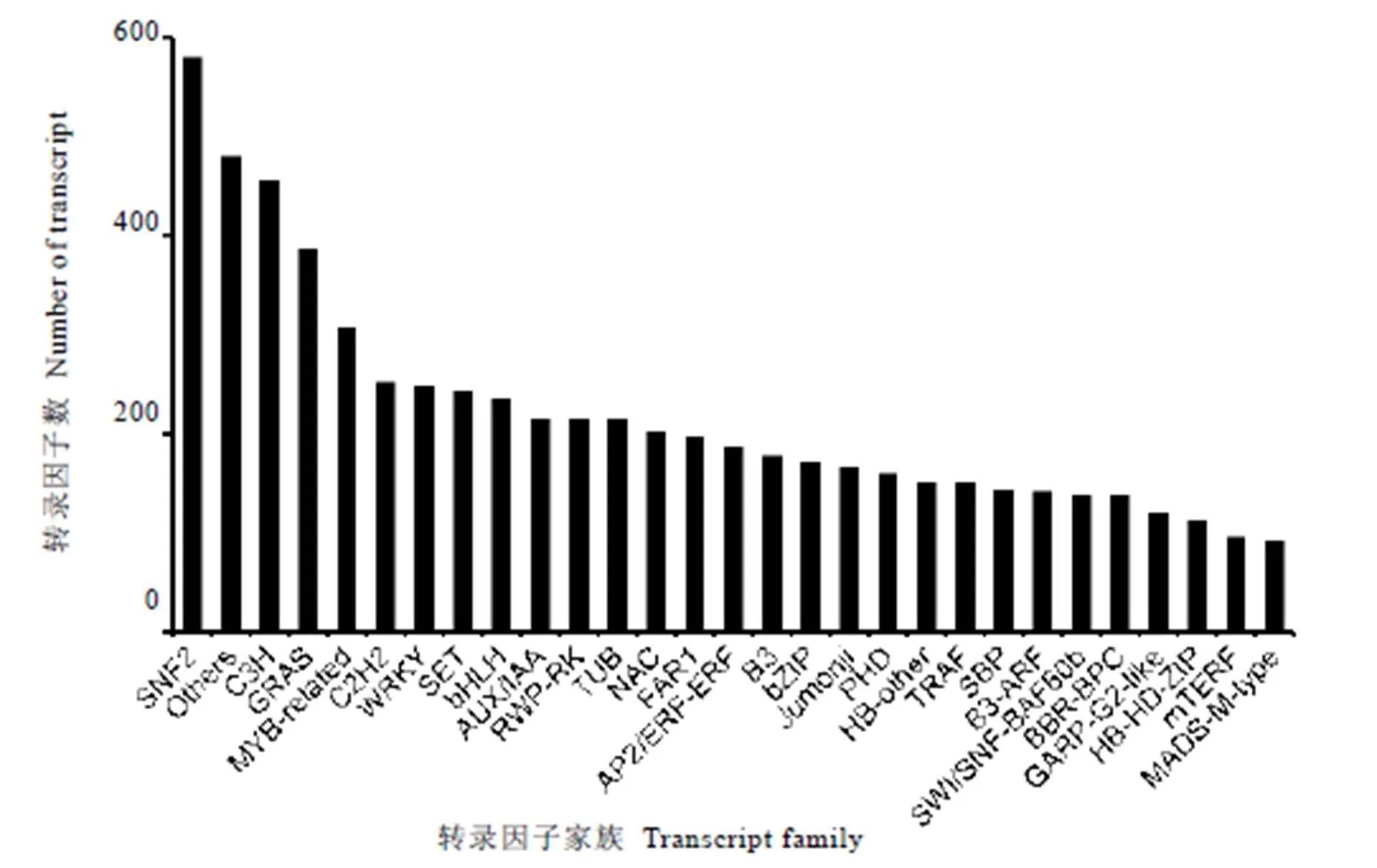
图2转录因子家族分析
茶树为叶用经济作物,云茶1号茶树其芽叶大,产量高,抗茶饼病和抗小绿叶蝉强,芽叶中儿茶素、黄酮以及氨基酸等生物活性物质含量高,制作的茶叶品质优,是一个集多种生物性状优异的茶树品种,是茶树遗传改良的重要亲本。本研究对云茶1号茶树全长转录组进行测序,通过7个数据库进行功能注释,在KEGG代谢通路注释到的105 972个unigenes,归属于216条通路。其中一些代谢途径与茶叶品质形成相关,包括氨基酸代谢(8 479个),咖啡因的代谢(41个),黄酮和黄酮醇的生物合成(86个),黄酮类化合物的生物合成(464个),单萜的合成(80个),倍半萜的生物合成(168个),二萜类化合物的生物合成(349),这些数据为云茶1号茶叶品质形成机制的研究提供了基础数据。还有一些与植物体内重要的抗逆途径相关,包括植物激素信号转导(1 772个),AMPK信号途径(1 149个),钙信号途径(431个),MAPK信号途径(1 194个),苯丙素的生物合成(826个)以及预测到的转录因子5 785个和所获得SSR信息,这将有助于进一步开展云茶1号茶树的抗病机理以及特异性状基因的标记性引物开发和遗传研究奠定基础。
[1] 张俊, 田易萍, 徐丕忠, 等. 优质、抗病大叶茶新品种“云茶1号”选育[J]. 茶叶, 2008, 34(1): 39-41.
[2] 王深. 云茶1号主要特点与栽培管理[J]. 农村实用技术, 2013(2):19.
[3] 冉隆珣, 玉香甩, 田易萍, 等. 不同茶树品种对茶饼病抗性鉴定初探[J]. 辽宁农业科学, 2017(1): 69-70.
[4] 田易萍, 张俊, 徐丕忠, 等. 茶新品种‘云茶1号’[J]. 园艺学报, 2009, 36(1): 153.
[5] Liao Y C, Lin S H, Lin H H. Completing bacterial genome assemblies: strategy and performance comparisons [J]. Scientific Reports, 2015, 5: 8747.
[6] Shin S C, Ahn D H, Kim S J, et al. Advantages of single-molecule real-time sequencing in high-GC content genomes [J]. PLoS One, 2013, 8(7): e68824.
[7] Gao J P, Wang D, Cao L Y, Sun H F. Transcriptome sequencing of codonopsis pilosula and identification of candidate genes involved in polysaccharide biosynthesis [J]. PLoS One, 2015, 10(2): 117-134.
[8] Shimizu K, Adachi J, Muraoka Y. ANGLE: a sequencing errors resistant program for predicting protein coding regions in unfinished cDNA [J]. Journal of Bioinformatics and Computational Biology, 2006, 4(3): 649-664.
[9] Zheng Y, Jiao C, Sun H, et al. iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases [J]. Molecular Plant, 2016, 9(12): 1667-1670.
[10] Lutoval A, Doduevai E, Lebedeva M A, et al. Transcription factors in developmental genetics and the evolution of higher plants [J]. Russian Journal of Genetics, 2015, 51(5): 449-466.
[11] 曹晨霞, 韩琬, 张和平. 第三代测序技术在微生物研究中的应用[J]. 微生物学通报, 2016, 43(10): 2269-2276.
[12] 任毅鹏, 张佳庆, 孙瑜, 等. 基于PacBio平台的全长转录组测序[J]. 科学通报, 2016, 61(11): 1250-1254.
[13] Shi C Y, Yang H, Wei C L, et al. Deep sequencing of thetranscriptome revealed candidate genes for major metabolic pathways of tea-specific compounds [J]. BMC Genomics, 2011, 12(1): 131.
[14] 陈林波, 夏丽飞, 周萌, 等. 基于RNA-Seq技术的“紫娟”茶树转录组分析[J]. 分子植物育种, 2015, 13(10): 2250-2255.
Full-length Transcriptome Analysis of Protected Cultivation ‘Yuncha 1’ (Var)
ZHU Xingzheng1,2, XIA Lifei1,2, CHEN Linbo1,2*, SUN Yunnan1,2, TIAN Yiping1,2, SONG Weixi1,2, JIANG Huibin1,2
1. Tea Research Institute, Yunnan Academy of Agricultural Sciences/Yunnan Technology Engineering Research Center of Tea Germplasm Innovation and Supporting Cultivation, Menghai, 666201, China; 2.Yunnan Provincial Key Laboratory of Tea Science, Menghai, 666201, China
To explore the genetic basis for important traits, the full-length transcriptome of the ‘Yuncha 1’ () was sequenced by using PacBio Platform. A total of 213 389 polished consensus were generated, 223 120coding sequences were predicted and annotated, and 195 062 SSR loci were found. According to NR databases, 170 264 homologous sequences were mapped to 980 species, 103 124 unigenes were further annotated and grouped into 26 functional categories in KOG databases, 65 524 unigenes were annotated against GO database and divided into cellular component, molecular function and biological process categories with a total of 55functional groups. KEGG pathway analysis showed that 105 972 unigenes could be broadly classified into 216 metabolism pathways according to their function, and some of them were involved in quality, bioactive substances, and resistance gene, etc. It is also predicted that there were 5 785 transcription factors belonging to 60 transcription factor families. The experimental results will give important data for development of SSRs of specifictraits,genetic analysis and studies involved in quality formation and resistence mechanism in tea cultivar ‘Yuncha 1’.
tea cultivar ‘Yuncha 1’, full-length transcriptome, gene analysis, function annotation
S571.1;S324
A
1000-369X(2018)02-193-09
2017-10-31
2017-11-27
国家自然科学基金项目(31660224、31560220、31460216)、云南省人才培养计划项目(2015HB105)
朱兴正,男,副研究员,主要从事茶树育种与生物技术方面的工作。
chenlinbo2002@sina.com
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
茶道(2022年3期)2022-04-27
中国生殖健康(2020年4期)2021-01-18
福建茶叶(2020年5期)2020-12-23
福建茶叶(2020年10期)2020-12-22
农技服务(2020年1期)2020-12-17
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
