
八聚(γ-氨丙基)倍半硅氧烷对芳纶纤维的抗紫外接枝改性
2018-04-04 06:41:53冒亚红
纺织科学与工程学报 2018年1期
管 宇,冒亚红
(1.中央军委后勤保障部军需军事代表局驻成都军事代表室,四川成都 610015;2.四川省纺织品生态染整高校重点实验室,四川成都 611731;3.成都纺织高等专科学校,四川成都 611731)
0 引言
芳纶以其高强、高模和耐高温特性,广泛应用于航空航天、国防、汽车工业、防护服、运动器材等方面,充分展示出它的优良性能[1-3]。但是,芳香族聚酰胺纤维容易受到紫外线影响,在紫外线辐射下,纤维很快出现变脆、力学性能下降的现象,颜色也由原来的黄色变成青铜色,化学稳定性变差[4-5],这些现象通常被称为光老化[6],严重限制了它的应用范围。
芳香族聚酰胺紫外光老化主要发生在大分子链上的酰胺键,紫外降解主要有两种方式。对于芳纶纤维来说,在无氧情况下,老化过程中主要发生的是Fries 重排和交联,大分子链一般不断裂;在有氧情况下,大分子链发生断裂,生成羧基等基团,这可以用自由基与氧的反应来得到较好的解释。本课题组利用八聚倍半硅氧烷(POSS)对紫外光选择性吸收的特性,将水溶性POSS盐酸盐用于芳纶和含杂环高性能纤维的抗紫外处理,处理后纤维的抗紫外性能得到显著提高[7-8],又利用Friedel-Crafts烷基化反应制备得到含POSS笼型结构的分散染料[9-11],采用该染料对上述纤维进行染色,做到了纤维着色与抗紫外改性同步实现。但是,前者处理后的纤维耐水洗性能较差,后者工艺流程较长,工艺条件难于控制。
本研究从同步提高对位芳纶(PPTA)纤维抗紫外性和耐水洗性的角度出发,利用氨基与酰氯反应生成酰胺键的原理,利用酸溶液对纤维表面进行适度刻蚀,将八聚(γ-氨丙基)倍半硅氧烷接枝在纤维表面,从而得到具有永久抗紫外性的芳纶纤维。
1 实验
1.1 材料和仪器
材料: 纤维直径11.8 μm 的对位芳纶1414 纤维(PPTA)(分别经过丙酮、乙醇浸泡和冷热水洗,以去除纺丝油剂,成都晨光化工研究院)。
药品:二氯甲烷、二氯亚砜、丙酮、丙酮、乙醇、硫酸、甲烷磺酸、多聚磷酸、氯化钠、无水硫酸钠和碳酸氢钠(均为分析纯,成都科龙化工试剂厂);γ-氨丙基三乙氧基硅烷(APTES)(湖北应城德邦化工新材料有限公司)。八聚(γ-氨丙基)倍半硅氧烷(氨基POSS)盐酸盐合成方法见参考文献7和8[7-8]。
仪器:YG001A 纤维电子强力测试仪(中国纺织研究院),FYH815A - II 垂直燃烧测试仪(温州方圆仪器有限公司),ICP-AES 测试仪(Thermo Fisher),S-250 扫描电子显微镜(Hitachi,Ltd,Japan),X射线衍射仪(D/maxIIIA,Japan),SOT-Ⅱ声速取向仪(山东莱州电子仪器厂),DCA-322 动力学接触角测试分析仪(Thermo,美国)。
1.2 预处理剂的筛选和纤维的预处理
本研究利用PPTA纤维轻度酸水解后表面出现的羧基,与氨基POSS的氨基进行共价键结合,以实现笼型POSS结构在纤维表面的固着。由于PPTA纤维的化学稳定性非常好,几乎对绝大多数强酸都有很好的稳定性[12-13]。早先的研究结果表明,仅有多聚磷酸、甲烷磺酸等少数几种强质子酸溶液能在常温下水解该纤维[12-14]。甲烷磺酸腐蚀性最高,纤维浸入后迅速结团成块,因此,甲烷磺酸不适合用于预处理。虽然多聚磷酸腐蚀性相对温和,但是其溶液的粘度相对较大,纤维在溶液中预处理时均匀性不好,因此,该酸溶液也不适用。硫酸为小分子无机强酸,低浓度溶液的腐蚀性也比较温和,即便是加热处理上述纤维,强力损失也比较小。根据以往的参考文献[12],加热后的硫酸溶液能够使上述纤维表面产生部分潜离子化,生成羧基、氨基或酰胺基等极性基团。因此,本研究选定硫酸作为预处理试剂。
将纤维浸泡在40℃~80℃的硫酸溶液(30%~70%)中2h~10h,取出后立即在自来水中冲洗10 min,而后放入5 g/L碳酸氢钠溶液浸泡5 min,以中和残留的酸,取出样品后再分别在冷、热蒸馏水中各清洗两遍,最后在避光通风环境中自然晾干。
1.3 纤维接枝改性
纤维酰氯化:将0.6 g酸预处理纤维称好置于圆底烧瓶中,加入15 mL二氯甲烷和10 mL二氯亚砜,加热至回流状态反应6 h。反应结束后,抽滤纤维,再用50 mL二氯甲烷搅拌清洗3次,捞出后悬挂于避光通风处晾干。
氨基POSS去除盐基:将4.0 g氨基POSS放入250 mL圆底烧瓶,加入20 mL二氯甲烷和10 mL饱和氯化钠水溶液。搅拌下缓慢滴加2 mol/L NaOH溶液,至水相pH达到9~10。瓶外用冰水浴控制温度,瓶内温度不高于5℃。而后分液,保留有机相,水相用20 mL二氯甲烷萃取3次,合并有机相。用无水硫酸钠干燥有机相,抽滤后旋干,得到粘性液体。
将氨基POSS的二氯甲烷溶液和酰氯化纤维放入500 mL圆底烧瓶,在加入适量二氯甲烷稀释,在氮气保护下,加热至回流状态反应6 h,再补加二氯亚砜2 mL,继续回流6 h。反应结束后,过滤取出纤维,再用20 mL二氯甲烷和20 mL丙酮分别搅拌清洗3次和1次,将得到的纤维在60℃下鼓风干燥6 h。方程式1是PPTA纤维与氨基POSS接枝改性反应历程。
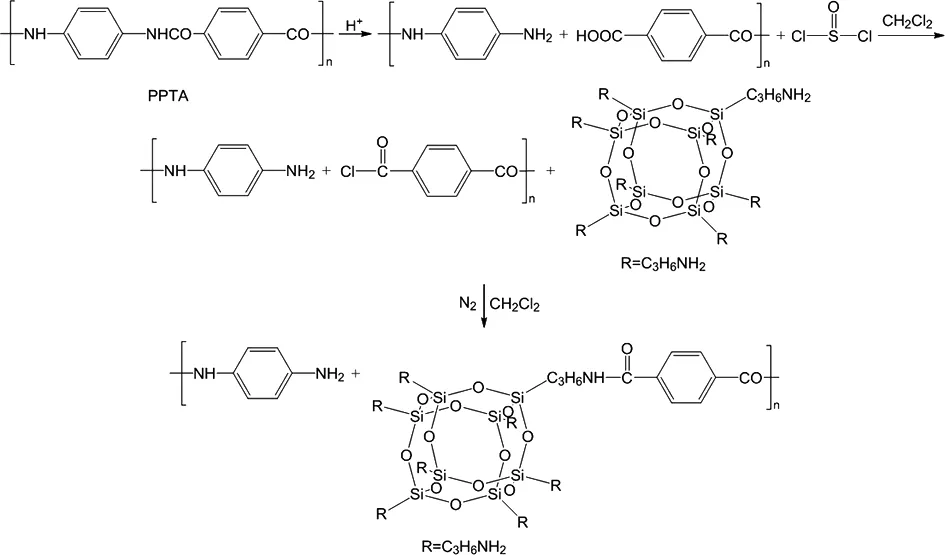
方程式1 PPTA纤维与氨基POSS接枝改性反应
1.4 纤维紫外加速老化实验
将处理后的纤维放在碘/镓灯(光源波长200nm~450 nm)进行照射,其辐照强度为800w/m2。试样距离光源35 cm,照射时由通风设备进行降温,实验温度保持在(30±0.5)℃。除文中特别指出,一般紫外加速老化时间为72 h。
1. 5 测试方法
1. 5. 1特性黏数[η]测定
以甲烷磺酸为溶剂,配制一定浓度的纤维溶液(浓度为(0.30~0.60)g/100mL),在(30±0.1)℃温度下,采用乌氏黏度计(毛细管直径为(0.9~1.0)mm)分别测定甲烷磺酸溶剂和样品溶液的流出时间,通过Huggins方程,导出试样的特性黏数[η][15]。
Huggins方程:ηsp/C=[η]+k[η]2C
(1)
式中ηsp=(t/t0)-1;[η]为特性黏数;t0为溶剂流出时间;t为纤维溶液流出时间;C为PBO溶液的浓度(g/dL)。
测定不同浓度C时的ηsp/C,然后将其对C作图得一直线,外推至 Y 轴上的截距即为特性黏度[η]。
1.5.2单纤强力测试
所有待测样品均在23℃、相对湿度68%条件下恒温恒湿保存24小时,根据《BISFA-2004聚合物纱线拉伸强力测试方法》在纤维电子强力测试仪上进行纤维强力测试。
1.5.3硅含量测试
采用电感耦合等离子体原子发射光谱法(ICP-AES)对处理后纤维的硅含量进行测试。将干燥纤维剪碎并精确称量0.100 g与0.8 g无水碳酸钠-硼酸混合物(质量比2:1)在干净的玻璃坩埚中充分研磨、搅拌、混合,并盖好坩埚盖。而后,将坩埚放入马弗炉,在900℃~950℃下灼烧12min~15min,让纤维充分灰化,取出后在通风条件下冷却至室温。然后,将灰分倒入100mL硝酸溶液(浓度20%),边加热,边搅拌,直至灰分全部溶解。最后,在200mL容量瓶中用去离子水进行稀释,制得测试溶液。硅含量测试在ICP-AES测试仪上进行。
1.5.4扫描电镜表征(SEM)
纤维样品表面喷金处理后,在扫描电子显微镜上观察纤维表面形貌的变化,加速电压为20KV[16]。
1.5.5X-射线衍射表征(XRD)
用X-射线衍射法分析酸预处理后纤维结晶变化情况。选用X-射线衍射仪进行XRD表征,测定波长为0.154nm的Cu放射X-射线,扫描衍射角2θ范围为5°~50°,测试样品被缠绕在不透明薄片上;为了防止透光,缠绕纤维分别沿两个垂直方向各缠绕一层;再使用MDI JADE 5.0软件对测试结果中出现的衍射峰进行面积计算,从而得到纤维的结晶度[17-18]。
1.5.6极限需氧指数
纤维燃烧实验根据《ASTM D2863-77塑料烛状燃烧最低需氧浓度测试方法(氧气指数)》用燃烧测试仪进行,在氧气-氮气混合气中,通过调节支持样品不间断燃烧30 s的氧气最低浓度,用(1)式计算得到LOI值[19-20]。
(2)
1.5.7纤维大分子取向度测定
样品测试前,在23℃、相对湿度68%条件下恒温恒湿保存24小时,而后用SOT-Ⅱ声速取向仪作为测试仪器,将纤维样品卷绕固定在声速仪两端,按选取10g张力夹夹持在待测纤维末端,测定20cm和40cm处声速值,同一样品测定10次取平均值[21-23]。
1.5.8表面自由能测试
表面自由能是指保持温度、压力和组成不变,增加单位表面积时,Gibbs自由能的增加值,也被称为表面Gibbs自由能,用符号γ表示,单位为J/m2。本研究中PBO纤维的表面自由能在DCA-322动力学接触角测试分析仪(Thermo,美国)上进行。目前纤维表面自由能采用接触角法进行计算,即通过测试纤维与不同极性液体(去离子水和二碘甲烷)的接触角结合Young’s方程和Owens-Wendt-Rabel and Kealble理论进行计算[24-25]。本研究测量表面自由能时,根据式(3~7)进行计算,计算中所涉及的基础数据见表1。
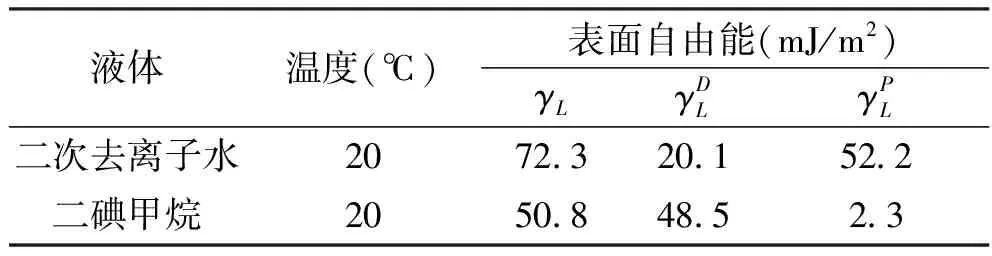
表1 两种液体的表面自由能[25]
(3)
(4)
(5)
(6)
(7)

2 结果与讨论
2.1 酸预处理工艺对纤维抗紫外性能的影响
为了给氨基POSS提供可反应的基团,本章采用轻度酸水解纤维的方法,使纤维大分子中的酰胺键少量断裂,使纤维表面生成供氨基接枝反应所需的羧基。影响硫酸预处理效果的主要因素有:酸浓度、预处理温度和预处理时间。
图1(a)是PPTA纤维经不同浓度硫酸溶液预处理后,采用1.3中酰氯化-氨基接枝反应得到的改性纤维,经过72 h紫外光加速老化,样品强力保留率的变化情况。图中曲线表明,随着浓度的提高,纤维的强力保留率逐步提高。由于纤维在酸溶液中的轻度水解作用,纤维表面生成了一定数量的羧基[14],为氨基POSS的共价键连接提供了反应点。随着酸浓度的提高,纤维表面的羧基数量逐步增加,接枝反应后纤维表面的POSS结构数量增加,因而抗紫外能力提高。当浓度增大到60%~70%时,纤维的强力保留率几乎保持不变,这说明纤维在硫酸溶液处理后,酸水解强力降低和抗紫外性能提高的平衡点已经出现,如果进一步提高酸浓度,样品强力保留率将会逐步下降。



图1酸溶液浓度、温度和处理时间对紫外照射样品强力保留率的影响
图1(b)中曲线显示,经紫外光加速老化后,氨基POSS接枝PPTA纤维随着预处理温度的上升,其强力保留率也随之上升。较高的处理温度,不仅能够促进水合氢离子的热运动,有利于其在纤维表面的吸附和向纤维内部的扩散,增强了纤维的溶胀和水解作用。一方面溶胀作用减弱了纤维大分子间的相互作用力,为水合氢离子进入纤维本体提供了通路;另一方面升温加快了酰胺键断裂,以至于大分子链上更加易于生成羧基。虽然,在本实验中随着温度的进一步上升,未出现强力保留率下降的现象,但是,纤维强力保留率的上升趋势已经变缓,如果采用无限制提高温度的办法促进水解反应,也将导致接枝改性纤维强力下降。由此可以推断,在该酸预处理工艺中,处理温度和酸溶液浓度两因素存在某种程度的等效性。
与很多有机物参加的化学反应相类似,PPTA纤维的水解过程也具有明显的时间依赖性。图1(c)中曲线表明,随着处理时间的延长,经紫外照射后纤维的强力保留率均随之上升,这表明硫酸溶液中的酸根离子和氢离子向纤维内部扩散需要一定时间,延长预处理时间使纤维溶胀和水解更加充分。这一点同提高酸溶液浓度和与预理温度的情况相类似,说明了这三个主要影响因素间存在某种程度的等效性,从物理化学角度讲,扩散过程是整个硫酸预处理工艺的动力学控制过程。在保证三种纤维抗紫外效果的基础上,充分考虑节能降耗和节约时间等因素,本文选取的酸预处理条件为:硫酸70%、80℃、10 h。
2.2 氨基POSS接枝改性后纤维中硅元素的含量
按照1.5.3方法,得到经氨基POSS接枝改性后PPTA纤维上硅元素的精确含量,并计算出每克纤维接枝POSS的含量,见表2。

表2 接枝后PPTA纤维上POSS的含量
表中数据表明,在氨基POSS接枝改性纤维中检测出含有硅元素,而未处理的纤维并不含有该元素,由此证明将氨基POSS用于酰氯化PPTA纤维接枝改性方法,以提高其抗紫外性能是切实可行的。通过与参考文献7数据对比[7],发现采用该方法改性后,单位质量纤维中所含的POSS含量略高于直接氨基POSS处理纤维。从理论上讲,纤维上POSS结构越多,纤维的抗紫外性能应该越好。但是,比较参考文献7中有关强力数据发现[7],接枝改性纤维的强力反而略微低于直接处理纤维。这可能是由于接枝过程中需要用硫酸溶液对纤维进行预处理,纤维表层遭到部分破坏和水解,致使纤维强力有所下降。同时,POSS结构主要吸附在氨基POSS直接处理纤维的表层,在加速老化时对紫外光的选择性吸收的效率比较高,而接枝改性纤维上的POSS除了固着在纤维表面以外,还有一部分接枝在结构较松散的纤维芯层,这部分POSS结构在紫外光照射时对纤维的保护作用非常有限,因而也会导致强力保留率偏低。
2.3 氨基POSS接枝改性对纤维溶液特性黏数的影响
在某些特性参数难以获得时,常把聚合物溶液的特性粘数用于表征聚合物的相对分子质量。采用改性纤维甲烷磺酸溶液的特性黏数变化,推断其相对分子质量的变化规律。表3是PPTA纤维经氨基POSS接枝改性前后和紫外光照射前后溶液特性黏数的变化情况。表中数据表明,纤维经过72 h紫外光照射后,特性黏数均有不同程度的降低,不过,接枝改性纤维降低的幅度较小。该现象说明纤维大分子链在紫外光照射后都发生了断裂,其聚合度降低。对于氨基POSS接枝改性纤维,由于纤维表层POSS结构对紫外光的选择性吸收作用,纤维大分子得到了一定程度的保护,其聚合度降低幅度减小,也从微观层面证明了采用该方法提高纤维抗紫外性能的有效性。

表3 氨基POSS接枝改性PPTA纤维溶液的特性黏数
与参考文献7中结果不同[7],接枝纤维溶液的特性黏数与原纤维比较接近,没有出现上升的现象,这可能是由于硫酸溶液预处理和酰氯化反应引起的聚合度降低(测试过程中以表观黏度的形式出现),部分抵消了POSS中氧原子与纤维大分子链上或甲烷磺酸上的氢原子发生氢键结合引起的表观黏度的上升。另外,将表中聚合物溶液特性黏数保留率与纤维强力保留率相比较(见2.1),发现接枝改性后纤维溶液的特性黏数保留率均高于强力保留率,这说明紫外光照射不仅仅使纤维大分子链发生断裂,更主要的是引起纤维超分子结构发生破坏,从而直接导致纤维强力的下降,下文中(见2.5)纤维的SEM照片将给出直接证据。这一点不同于由于染整加工不当造成的棉纤维的潜在损伤。
2.4 氨基POSS接枝改性对纤维特征基团的影响
图2是PPTA纤维原样、硫酸溶液处理样和氨基POSS接枝改性样的红外光谱曲线。图中曲线显示,处理前后纤维的红外光谱整体变化不大,而且吸收曲线上1633.41 cm-1对应的酰胺键C=O伸缩振动吸收峰没有发生改变[26],由此说明,处理工艺对纤维本身官能团的影响不大,纤维微观结构保持较好。

图2 氨基POSS接枝改性PPTA纤维的红外光谱
图中,接枝处理后PPTA纤维分别在红外谱线的1108.87 cm-1比原有吸收峰的强度有所提高。可能的解释是,由于Si-O-Si在1030cm-1~1110cm-1有较强的伸缩振动,与纤维附近的吸收峰发生重叠,因而,未显示出新的独立的吸收峰,仅仅增强了原有吸收峰的吸收强度。同时,纤维在3300cm-1附近吸收峰的强度有所提高,从而直接证明了纤维表面羧基数量的增加。
2.5 氨基POSS接枝改性对纤维表面形态的影响
图3是PPTA纤维经硫酸溶液预处理和氨基POSS接枝改性以及紫外光照射后的表面形态变化情况。将图中(a)和(b)与参考文献7中原纤维SEM照片相比[7],除了接枝改性后纤维表面出现轻微轴向条痕或少量表皮鼓起外,纤维的表面形态几乎没有改变,这说明氨基POSS接枝改性对纤维皮层结构的损伤非常有限,也印证本文2.1中了改性纤维强力损失小的实验结果。

注:(a)硫酸处理样;(b)接枝改性样;(c)紫外照射改性样。
图3PPTA纤维表面SEM照片
再将图(c)与参考文献7中相关SEM照片进行对比[7],氨基POSS接枝改性纤维在紫外光照射后的表面形态保持情况明显好于未处理和氨基POSS直接处理纤维,不但没有出现表皮脱落和原纤化现象,而且纤维表面非常光滑。这一方面可能是因为低浓度的硫酸溶液很难对纤维皮层进行刻蚀,仅仅能够溶解纤维纺丝过程中残留在纤维表面的微原纤,从而纤维表面变得更加光滑;另一方面由于纤维经过长时间酸溶液浸泡,纤维表面的氨基POSS固着点,也就是水解出现的羧基,分布比较均匀,接枝后POSS结构在纤维上分布也更加均匀,从而在紫外光照射时对纤维有更好的保护作用。
2.6 氨基POSS接枝改性对纤维超分子结构的影响
声速法是利用声波在不同大分子链中传播速率的不同,来判断大分子链的相对取向程度,是测量纤维取向度的一种常用方法。当声音沿着纤维的轴向传播时,速率最大,而沿径向传播时,速度最小。因此,对于同种纤维而言,较高的传播速度则对应着较高的取向度[21-23]。表4 是PPTA纤维接枝改性和紫外光照射前后,声波在纤维中传播速度的变化情况。表中数据表明纤维经紫外光照射后,纤维中声波传播速率均有不同程度的降低,尤其以未经氨基POSS改性纤维更为明显。这可能是因为紫外光与空气的共同作用致使纤维大分子链断裂,纤维晶区被部分破坏,纤维非晶区比例提高,从而降低了声波的传递速率。纤维改性后,部分紫外光被吸收屏蔽,纤维得到一定程度的保护,纤维损伤程度也有所降低,因而,POSS改性后纤维的声波传递速率降低比较有限,从微观角度证实了该处理工艺对纤维大分子链的排列有序度影响不大。

表4 接枝改性工艺对PPTA纤维取向度的影响
图4是纤维氨基POSS接枝改性和紫外光照射前后的XRD曲线,比较图中四条曲线,接枝改性和紫外光照射都没有使样品的衍射角发生明显变化,表明纤维的结晶形态没有发生变化,但是衍射峰的强度有不同程度的减弱。采用MDI Jade5.0软件对每条衍射曲线的峰下面积进行积分计算,得到纤维的结晶度参数,见表5。

图4 PPTA纤维XRD曲线

纤维结晶度(%)未经紫外照射紫外照射72h结晶度损失率(%)原样731408323接枝改性样58453977
表5数据显示,PPTA纤维接枝改性后结晶度有少量降低,说明硫酸预处理和接枝改性工艺虽然会部分破坏纤维表面的结晶结构,但是这种破坏作用是十分有限的,纤维皮层和主体的结构基本没有发生明显变化,这一点从纤维表面SEM照片中已经被观察到。然而,紫外照射后,纤维结晶度均有不同程度的降低,但是接枝改性试样的下降幅度明显小于未改性试样。结合纤维表面SEM照片和拉伸强力数据,纤维拉伸强力与纤维皮层结构是否完好关系密切,该接枝改性工艺能够在紫外光照射时很好的保护纤维表面,使纤维皮层的结晶部分得以保持,也就是结晶度下降较少,从而也获得了较好的力学性能。
2.7 氨基POSS接枝改性对纤维热性能的影响
图5(a)是PPTA纤维热分解时的TG曲线,对该曲线进行微分计算后,得到DTG曲线,如图5(b),另纤维的热重法分析结果见表6。表中数据显示,经氨基POSS接枝改性的PPTA纤维,无论是否经过紫外光照射,热分解初期(失重5%)的边界温度均高于未处理纤维。
这一阶段的重量损失主要来源于纤维合成和纺丝过程中混入的小分子溶剂或油剂的挥发,还包括纤维合成中未与大分子链相连的齐聚物,以及紫外光照射引起的聚合物大分子断链产生的分子链段发生热降解。由于纤维表层引入大量热稳定性较高的POSS结构,虽然它不能阻止小分子溶剂或油剂的挥发,但是它却能够有效延缓聚合物中的齐聚物或聚合度较低的分子链段的热降解,从而提高了初始热降解温度。而且这种热失重的差异对于经紫外光照射后的纤维更加显著,这也就进一步证明了上述的推论。

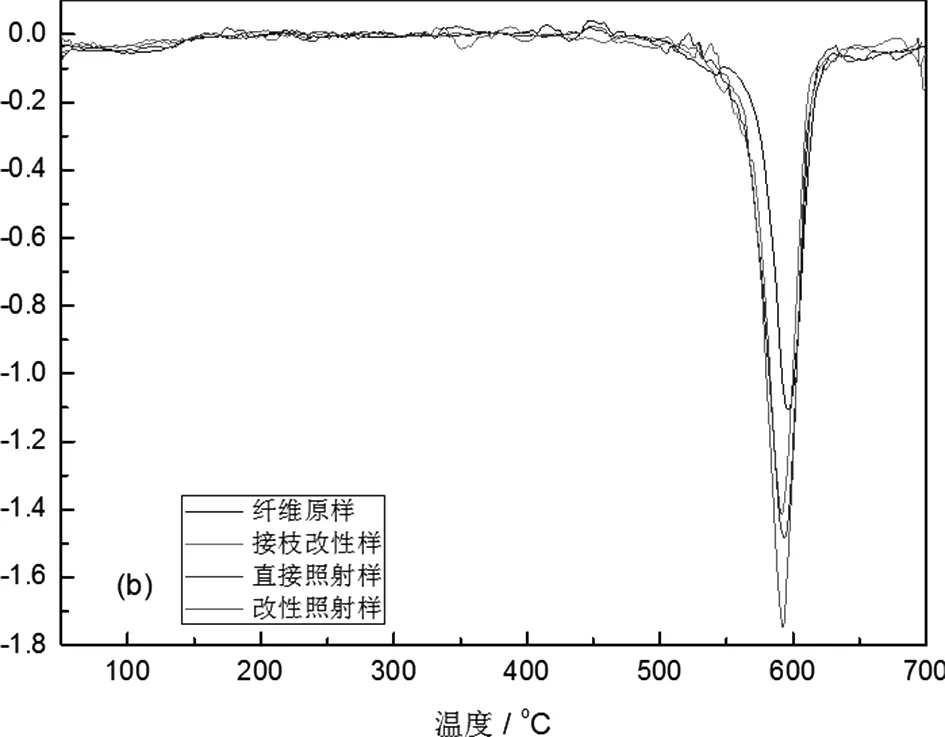
图5 PPTA纤维的TG和DTG曲线
另外,通过比较残渣残留率发现,改性后纤维无论是否经过紫外光照射,其数值均有降低,这可能是因为接枝改性纤维需要经过硫酸溶液预处理,纤维本体或多或少都会受到影响,原本需要在更高温度才分解炭化的结晶态聚合物,在加热前就遭到一定破坏,从而在较低的温度就被分解,从而残渣较少。因此,该接枝改性工艺对提高PPTA纤维的阻燃性效果不佳。
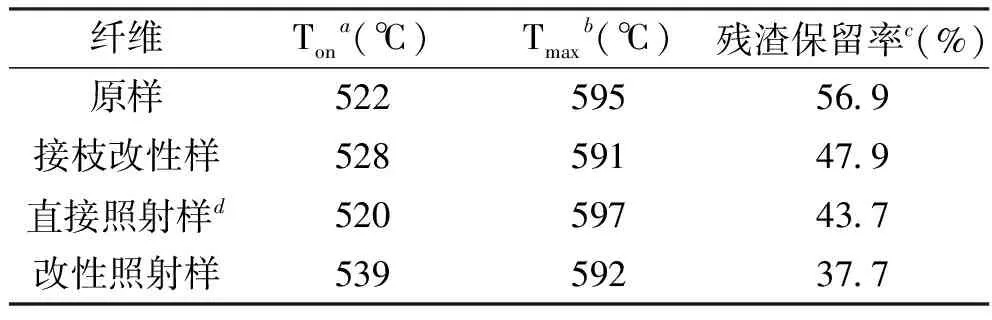
表6 PPTA纤维TG曲线相关数据
注:aTon表示质量(损失5%)的边界温度;bTmax表示最快热分解温度;c测试温度为700℃;d紫外光照射时间为72 h。
另外,表中数据还显示,接枝改性PPTA纤维的最快热分解温度(Tmax)下降不显著,虽然接枝改性工艺对纤维本身有一定破坏,导致Tmax降低,但是在酸处理时或多或少会残留一些硫酸,在氮气氛中加热纤维,硫酸具有一定的阻燃作用,因此,其最快分解温度变化不明显。

表7 接枝改性工艺对PPTA纤维极限需氧指数(LOI)的影响
极限氧指数(LOI)是指在规定的条件下,材料在氧氮混合气流中进行有焰燃烧所需的最低氧浓度,是常被用于表征聚合物燃烧性能和判断物质阻燃性能的一种定量参数[19]。表7数据显示,改性后PPTA纤维的LOI值几乎没有变化,这与氨基POSS直接处理纤维的情况是完全不同的[8]。可能的解释是,硫酸溶液预处理和接枝改性反应使纤维结构变得蓬松,增加了纤维的可燃性,这一点与紫外光加速老化后纤维的情况相类似。再加之,残留在纤维中的硫酸在氧气氛燃烧过程中,有促进氧化的作用,因而导致了LOI值上升幅度的降低。同时,经两种不同的氨基POSS处理工艺得到纤维试样在阻燃性能的差异,也是它们微观结构差异的宏观表现。
2.8 氨基POSS接枝改性对纤维表面性能的影响
采用表面自由能考察氨基POSS接枝改性纤维表面亲水或疏水性能的变化[24, 27, 28]。表8是氨基POSS接枝改性后PPTA纤维接触角与表面自由能的变化情况。表中数据显示,改性纤维与水的接触角明显减小,而与非极性二碘甲烷的接触角显著增大,同时纤维的表面自由能也同步大幅提高,这主要是因为固着在纤维表面的氨基POSS增强了纤维表面的极性。一方面,离子化的氨基能够与水分子形成稳定的水合离子;另一方面,POSS中的氧原子也能与水分子中的氢原子形成氢键,提高分子间的亲和力。

表8 接枝改性工艺对PPTA纤维接触角和表面自由能的影响
2.9 接枝改性纤维抗紫外性能耐久性
参考文献7和8采用的POSS改性纤维以提高其抗紫外性能,都是通过选用特定结构的POSS分子,利用它们与纤维间的物理作用力(范德华力),将POSS结构固着在纤维上[7, 8]。但是,分子间范德华力的作用强度相对于共价键而言,要低至少一个数量级[29],因此,为了使POSS结构更加坚牢的固着在纤维上,使纤维获得永久的抗紫外性能,从而提出了本方法,这也是该工艺的最大特点。
为了掌握氨基POSS直接改性和接枝改性两种工艺处理后纤维抗紫外耐久性间的差异,对两种处理试样分别进行2至10次水洗,对水洗后纤维再进行72 h紫外光照射,得到的纤维强力保留率见图6。
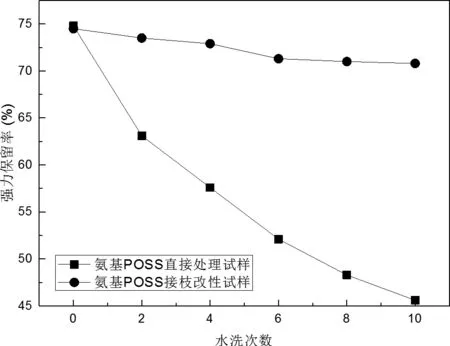
图6 氨基POSS处理和接枝改性纤维抗紫外耐久性
图中曲线表明,对于直接处理试样,随着水洗次数的增加,纤维的强力保留率大幅下降,尤其在前4次水洗过程中,纤维的强力保留率几乎下降了20%。由于本研究使用的八聚氨基POSS盐酸盐的水溶性特别好,在加工过程中可以根据需要配置各种浓度的处理液,但是,处理试样上的氨基POSS也很容易在水洗过程中,从纤维表面脱落,使纤维的抗紫外性能迅速下降。对于接枝改性试样,经过10次水洗,其强力保留率下降微乎其微(不足2.5%),这主要归功于纤维与POSS间的键能较高的共价键。即便是发生了微小的强力损失,也主要发生在多次水洗以后(6次以后),这可能是反复浸泡弱碱性洗涤液,造成了POSS与纤维间的共价键水解所致。
3 结论
利用氨基与酰氯反应生成酰胺键的原理,采用硫酸预处理和酰氯化反应,得到带有酰氯基的PPTA纤维,通过与氨基POSS反应,使对紫外光有选择性吸收的八聚氨基POSS通过酰胺键与纤维连接,从而提高了纤维的抗紫外性能。在适当条件下,硫酸浓度增加、温度提高和时间延长都有利于加快水合氢离子的热运动,增进纤维溶胀,促进水解反应的进行,在纤维表面生成供氨基POSS共价键结合的固着点,使POSS结构在纤维上固着成为可能,使经过紫外光照射后纤维的强力保留率明显上升。通过对接枝改性后和紫外光照射后纤维的结构表征和性能测试,发现该接枝改性工艺不仅较好保持了纤维的基本结构和热稳定性,而且提高了纤维的表面润湿性,对制作纤维复合材料更加有利。经紫外光照射后,接枝改性纤维的表面形态、超分子结构、大分子聚合度的保持明显好于未改性纤维。同时,相对于氨基POSS直接处理纤维,接枝改性纤维的抗紫外耐久性更好,揭示了氨基POSS与纤维共价键连接的优越性,证实了该方法更适用于聚芳类纤维永久性抗紫外改性。
[1]孙晓婷, 郭亚. 芳纶纤维的研究现状及应用 [J]. 成都纺织高等专科学校学报, 2016, 33(3): 164-168.
[2]凌新龙, 郭立富, 黄继伟, 杨春霞. 芳纶纤维表面改性技术研究现状与进展 [J]. 成都纺织高等专科学校学报, 2016, 33(2): 44-52.
[3]张文惠, 徐广标. 芳纶与PTFE基布针刺非织造布结构与透气性能的测试评价 [J]. 成都纺织高等专科学校学报, 2015, 32(4): 72-74.
[4]Carlsson D J, Gan L H, Parnell R D, Wiles D M. The Photodegradation of Poly(1,3-Phenylene Isophthalamide) Films in Air [J]. Journal of Polymer Science: Polymer Letters Edition, 1973, 11(11): 683-688.
[5]Johnson L D, Tincher W C, Bach H C. Photodegradative Wavelength Dependence of Thermally Resistant Organic Polymers [J]. Journal of Applied Polymer Science, 1969, 13(9): 1825-1832.
[6]Hargreaves G, Bowen J H. Combined Effects of Gamma and Ultraviolet Radiation Plus Heat on Fibrous Polyamides [J]. Textile Research Journal, 1973, 43(10): 568-576.
[7]Mao Y, Zhou W, Xu J. Ultraviolet Resistance Modification of Poly(P-Phenylene-1,3,4-Oxadiazole) and Poly(P-Phenylene Terephthalamide) Fibers with Polyhedral Oligomeric Silsesquioxane [J]. Journal of Applied Polymer Science, 2015, 132(41): n/a-n/a.
[8]冒亚红, 管宇. 八聚(Γ-氨丙基)倍半硅氧烷盐酸盐对pbo纤维抗紫外改性 [J]. 成都纺织高等专科学校学报, 2017, 34(1): 36-44.
[9]Mao Y, Song Q, Guan Y. Improving the Uv Resistance of Aromatic Poly(L,3,4-Oxadiazole) Fiber Using a Disperse Dye Modified with Octavinyl POSS. Part 1: Preparation of Dye and Dyeing [J]. Journal of Applied Polymer Science, 2017, 134(16): n/a-n/a.
[10] Mao Y, Zheng X, Guan Y. Improving the Uv Resistance of Aromatic Poly(1,3,4-Oxadiazole) Fiber Using the Disperse Dye Modified with Octavinyl POSS. Part 2: Fixing Form of Dye and Uv Resistance Ability [J]. Journal of Applied Polymer Science, 2017, 134(23): n/a-n/a.
[11] 管宇, 冒亚红. 八聚乙烯基倍半硅氧烷改性分散染料对芳纶纤维抗紫外染色 [J]. 成都纺织高等专科学校学报, 2017, 34(1): 13-20.
[12] Forster A L, Pintus P, Messin G H R, Riley M A, Petit S, Rossiter W, Chin J, Rice K D. Hydrolytic Stability of Polybenzobisoxazole and Polyterephthalamide Body Armor [J]. Polymer Degradation and Stability, 2011, 96(2): 247-254.
[13] Morgan R, Butler N. Hydrolytic Degradation Mechanism of Kevlar 49 Fibers When Dissolved in Sulfuric Acid [J]. Polymer Bulletin (Berlin), 1992, 27(6): 689-696.
[14] Chatzi E G, Tidrick S L, Koenig J L. Characterization of the Surface Hydrolysis of Kevlar-49 Fibers by Diffuse Reflectance Ftir Spectroscopy [J]. Journal of Polymer Science Part B: Polymer Physics, 1988, 26(8): 1585-1593.
[15] Gomes D, Borges C P, Pinto J C. Evaluation of Parameter Uncertainties During the Determination of the Intrinsic Viscosity of Polymer Solutions [J]. Polymer, 2000, 41(14): 5531-5534.
[16] Qiao X, Li W, Sun K, Xu S, Chen X. Nonisothermal Crystallization Behaviors of Silk-Fibroin-Fiber-Reinforced Poly(ε-Caprolactone) Biocomposites [J]. Journal of Applied Polymer Science, 2009, 111(6): 2908-2916.
[17] 张超, 曾黎明, 陈雷, 赵军. Pbo纤维及其改性的研究进展 [J]. 合成纤维工业, 2010, 33(3): 49-52.
[18] Tashiro K, Kobayashi M. Theoretical Young′s Moduli of Poly(P-Phenylenebenzobisthiazole) and Poly(P-Phenylenebenzobisoxazole) [J]. Macromolecules, 1991, 24(12): 3706-3708.
[19] Borah J, Karak N. Blends of Triazine-Based Hyperbranched Polyether with Ldpe and Plasticized Pvc [J]. Journal of Applied Polymer Science, 2007, 104(1): 648-654.
[20] Nelson M I. A Dynamical Systems Model of the Limiting Oxygen Index Test: Ii. Retardancy Due to Char Formation and Addition of Inert Fillers [J]. Combustion Theory and Modelling, 2001, 5(1): 59-83.
[21] 邓澧儒, 钱人元. 声速法测定纤维的取向度 [J]. 高分子通讯, 1964, 6(2): 117-123.
[22] Moseley W W. The Measurement of Molecular Orientation in Fibers by Acoustic Methods [J]. Journal of Applied Polymer Science, 1960, 3(9): 266-276.
[23] Charch W H, Moseley W W. Structure-Property Relationships in Synthetic Fibers: Part I: Structure as Revealed by Sonic Observations [J]. Textile Research Journal, 1959, 29(7): 525-535.
[24] 徐广标, 杨立荣. 3种天然纤维表面自由能计算与评价 [J]. 东华大学学报(自然科学版), 2013, 39(3): 280-282.
[25] Chwastiak S. A Wicking Method for Measuring Wetting Properties of Carbon Yarns [J]. Journal of Colloid and Interface Science, 1973, 42(2): 298-309.
[26] 陆赵情, 刘俊华, 张美云, 刘俊. 对位芳纶纤维物化结构的研究 [J]. 纸和造纸, 2013, 32(6).
[27] 张世举, 程延海, 邢方方, 李梦晗, 朱真才. 接触角与表面自由能的研究现状与展望 [J]. 煤矿机械, 2011(10): 8-10.
[28] 朱埗瑶. 表面与表面自由能 [J]. 大学化学, 1987, 2(4): 23-25.
[29] 孙铠, 蔡再生. 染整工艺原理(第三册) [M]. 北京: 中国纺织出版社, 2010.
猜你喜欢
四川劳动保障(2021年9期)2022-01-18 05:11:30
合成树脂及塑料(2020年6期)2020-12-29 07:02:02
四川冶金(2019年5期)2019-12-23 09:04:48
今日农业(2019年15期)2019-09-03 01:08:34
石油沥青(2019年4期)2019-09-02 01:41:54
小哥白尼(军事科学)(2018年2期)2018-05-25 03:12:52
中国塑料(2016年3期)2016-06-15 20:30:03
电线电缆(2016年5期)2016-02-27 09:02:08
中国塑料(2015年1期)2015-10-14 00:58:41
中国塑料(2014年1期)2014-10-17 02:46:36
