
Heterozygous carriers of galactocerebrosidase mutations that cause Krabbe disease have impaired microglial function and defective repair of myelin damage
2018-04-04 07:40NicoleScottHewittChristopherFoltsMarkNoble
中国神经再生研究(英文版) 2018年3期
Nicole J. Scott-Hewitt, Christopher J. Folts, Mark D. Noble
Department of Biomedical Genetics, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA
Introduction
Although it is widely recognized that being a heterozygous carrier of mutations in a wide range of genes can increase the likelihood of developing a variety of different diseases,our understanding of the biological foundations underlying such increased vulnerability is greatly lacking.
One example of this problem is seen in families of children with Krabbe disease (KD), a severe lysosomal storage disorder (LSD) caused by mutations in the enzyme galactocerebrosidase (GALC). Children with KD, who carry mutations in each copy of the GALC gene (Wenger et al., 1974,1997, 2000), develop a severe neurodegenerative disease characterized by extensive damage to myelin (the lipid-rich insulating material critical for neuronal conduction in both the central nervous system (CNS) and the peripheral nervous system (PNS)). In severe forms of KD, nervous system damage is so extensive as to typically cause death within 2–3 years after birth (Suzuki, 2003).
Although it has long been assumed that heterozygous carriers of mutations able to cause KD (which include all parents of children with KD) are biologically normal, it is increasingly apparent that this is not correct. Although this research is in its early stages, it is already clear that carriers of mutations that cause KD are at increased risk for development of open angle glaucoma (Liu et al., 2011) and also for pulmonary artery enlargement in association with chronic obstructive pulmonary disease (Lee et al., 2015). Such mutations also have been suggested to be of possible interest in late-onset synucleinopathies (Marshall and Bongarzone, 2016).
A similar, and more extensively studied, situation is also seen in the case of Gaucher disease (GD, another severe LSD). Heterozygosity for variants in glucocerebrosidase(GBA, the enzyme mutated in GD) is one of the strongest genetic risk factors for Parkinson’s disease (PD), and GBA mutations are found with even more frequency in the PD population than mutations in genes associated with familial forms of this disease (e.g., Neumann et al., 2009; Schapira,2015; O’Regan et al., 2017). Moreover, PD patients with GBA variants tend to have earlier onset of disease symptoms and more severe cognitive decline when compared to PD patients that do not harbor GBA variants. While the genetic link between GBA and PD is well established, the underlying biological mechanisms are only beginning to be understood.
The increased disease vulnerability for heterozygous carriers of mutations that cause KD or GD affects the parents and families of afflicted children, of course, but is also more broadly relevant: Although the frequency of KD and GD is very low (1:80,000–100,000), the gene frequency for heterozygous carriers of disease-causing mutations is high enough (1:125–150) (Wenger, 1993) as to represent a significant proportion of the population. Just these two lysosomal storage disorders alone provide a predicted incidence of disease-relevant mutations in the population of ~1:60 to 1:75.Moreover, there are over 40 different lysosomal storage disorders for which information on associated disease risks has not yet been determined but which also may confer similar vulnerabilities.
One example of the potential relevance of variation in genes that cause KD to the general population comes from identi fication of genetic loci that increase the risk of developing multiple sclerosis (MS). MS is the most common autoimmune disorder affecting the CNS, and is associated with widespread damage to myelin and eventual failure to repair such damage, with multiple neurological sequelae (Sawcer et al., 2011, 2014; George et al., 2016). It has long been recognized that there are genetic factors that increase the likelihood of an individual developing MS. For example, it has long been known that there is a strong association with the human leukocyte antigen (HLA)-DRB1 locus (a group of genes that serve as the major histocompatibility complex(MHC) (Barcellos et al., 2006)). More recent studies have identi fied over 100 associated common variants that also increase the risk for development of MS (Sawcer et al., 2014).Some of the genetic loci associated with an increased risk of developing MS are known to be involved in immunological activities, and have often been associated with other autoimmune disorders (Sawcer et al., 2011, 2014). For many others,however, any potential biological linkage to MS remains obscure (Bashinskaya et al., 2015; Jokubaitis and Butzkueven,2016).
We were particularly interested in findings that one of the genetic risk factors for MS maps to the GALC locus(Sawcer et al., 2011, 2014). The GALC SNP, rs74796499,was one of the strongest vulnerability loci identi fied in the genome-wide association study (GWAS) study conducted by the International Multiple Sclerosis Genetics Consortium and the Wellcome Trust Case Control Consortium, and was one of the few loci in this study to show an odds ratio > 1.2 and an allele frequency close to 1 (Sawcer et al., 2011, 2014).This SNP is predicted to lead to a splice-region variant in the GALC gene (Sawcer et al., 2014), and while it remains unknown if the variant identi fied by the MS GWAS is associated with KD, intronic variants have been described in several late-onset KD patients (Kukita et al., 1997), and variants leading to altered mRNA splicing are also known to cause disease (Tappino et al., 2010).
The observations discussed in this introduction lead to two questions: First, how does being a heterozygous carrier of KD-causing mutations in GALC increase the risk of any disease? Second, what are the biological mechanisms that may enable variants in the GALC locus to increase the risk of developing MS? More broadly, these concerns could be phrased as asking what is the underlying biology that makes a genetic variant a risk factor?
Mice heterozygous for loss-of-function mutation in the GALC gene have normal levels of myelin and do not show increased myelin damage in response to cuprizone exposure
Two possible means by which mutations in GALC could increase the risk for developing MS would be to cause subclinical damage to myelin and/or to increase the amount of damage caused by demyelinating insults. To determine whether heterozygous loss of function in the GALC gene can cause or exacerbate myelin damage we examined unmanipulated twitcher (twi) heterozygotes (GALC+/–) and wild-type(WT) littermates and also examined mice exposed to cuprizone, a copper-chelating compound, in their food for four weeks. Cuprizone exposure is one of the most widely studied models of myelin damage (e.g., Kipp et al., 2009; Gudi et al., 2014; Praet et al., 2014) and a four-week exposure period causes extensive myelin loss in the corpus callosum (the major myelinated tract of the rodent CNS) (Matsushima and Morell, 2001). Following removal of cuprizone from the diet,remyelination then occurs. This is characterized by an initial increase in the recruitment and proliferation of the cells that give rise to myelin-forming oligodendrocytes, the oligodendrocyte/type-2 astrocyte progenitor cells (also referred to as oligodendrocyte precursor cells, and here abbreviated as O2A/OPCs). After proliferation, these progenitor cells then differentiate into oligodendrocytes, which go on to generate new myelin. Cuprizone exposure is also accompanied by increases in neuroin flammation (Gudi et al., 2014).
We first observed that unmanipulated WT and GALC+/–age-matched animals did not exhibit any differences in amounts of corpus callosal myelin, oligodendrocytes or proliferating O2A/OPCs, and no differences in motor behavior. There were no detectable differences in callosal myelin(as measured by FluoroMyelin™staining), in numbers of oligodendrocytes (defined as Olig2/glutathione-S-transferaseπ double-positive cells) or in numbers of proliferating O2A/OPCs (defined as Olig2/Ki67 double-positive cells).In addition, there were no gross changes in motor function as examined by analysis of several gait parameters that are altered in GALC–/–animals (Folts et al., 2016). Moreover,even though GALC+/–mice only have half the GALC enzyme activity of WT mice, they also have normal levels of psychosine, the toxic lipid that accumulates in KD and that disrupts multiple lysosomal and cellular functions (see Folts et al., 2016 and references therein).
Due to the extensive myelin damage that occurs in twi homozygotes, our expectations were that GALC+/–mice exposed to cuprizone for four weeks would show an increase in the amount of demyelination caused, but this prediction turned out to be incorrect. Two-month-old WT and GALC+/–littermates exposed to cuprizone-containing chow for 4 weeks were similar in the extent of myelin damage observed, and also in weight loss during cuprizone exposure and in weight gain after removal from cuprizone chow.

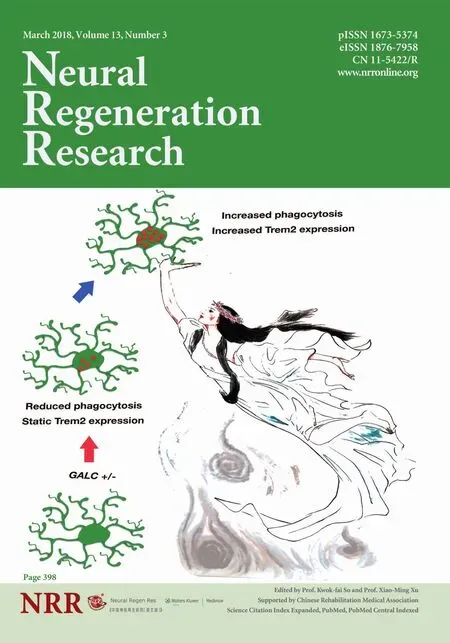
Figure 1 Summary of differences between wild-type (WT) mice (A) and unmanipulated twitcher (twi) heterozygotes (GALC+/–) mice (B)exposed to cuprizone for four weeks.
Heterozygous GALC+/– mice are defective in repair of cuprizone-induced myelin damage
Where GALC+/–mice and their WT littermates did differ,however, was in their ability to repair the damage caused by exposure to cuprizone (as summarized inFigure 1). When examined at 2-weeks after restoration to a normal (cuprizone-free) diet, the GALC+/–mice showed signi ficantly less myelin staining when compared to recovery-matched WT littermates. This defect in remyelination was present for at least 4 weeks of recovery.
Differences in the remyelination response were also seen at the cellular level, for both oligodendrocytes and dividing progenitor cells. Although GALC+/–and WT mice showed similar reductions in oligodendrocyte numbers when examined immediately following 4 weeks of cuprizone exposure,the GALC+/–mice had signi ficantly fewer oligodendrocytes during recovery as compared with recovery-matched WT littermates. GALC+/–mice also showed a signi ficant reduction in O2A/OPC numbers when compared to recovery-matched WT littermates at 2 and 3 weeks after restoration to a cuprizone-free diet. Such a difference was not seen in mice at the completion of the cuprizone treatment, and seemed instead to be speci fic to the repair period.
Heterozygous loss of GALC function causes an impaired microglial response to myelin damage
Alterations in the response to myelin damage also were seen in clearance of myelin debris. Such clearance is necessary for repair following a demyelinating injury, and delayed clearance of debris can prevent proper O2A/OPC differentiation and subsequent remyelination (Kotter et al., 2006; Kuhlmann, 2008; Lampron et al., 2015). When we stained brains of treated mice with an antibody that speci fically recognizes damaged myelin basic protein (dMBP; Matsuo et al., 1997;Ihara et al., 2010), we found that GALC+/–animals showed significantly more dMBP staining than their WT counterparts at both 2- and 3-week recovery. This difference was seen even though staining for dMBP did not differ between GALC+/–and WT mice examined promptly at the end of 4 weeks of cuprizone exposure, and thus was a characteristic of the repair phase rather than the damage phase.
Our further studies suggested that an important contributor to the failure to clear myelin debris may be a compromised function of microglia, which are thought to be the primary phagocytic cell responsible for contributing to remyelination after cuprizone injury (e.g., Napoli and Neumann, 2010; Voss et al., 2012; Gudi et al., 2014; Praet et al.,2014; Lampron et al., 2015). As with our examination of myelin damage, changes in the microglial response appeared to be speci fic to the repair phase after cuprizone was removed from the diet. Microglial numbers and morphology were similar in WT and GALC+/–mice examined immediately after cuprizone exposure (as determined by staining with Iba-1, a microglia speci fic marker (Ito et al., 1998)), both in numbers of microglia and in their morphology. In contrast,when we examined the CNS at 4 weeks post-recovery, we saw a more rapid return to baseline for numbers of microglia in mutant mice. We also observed that the microglia in WT mice appeared more ramified, with increased process complexity, while the GALC+/–microglia appeared to have stunted, less complex processes.
In further studying microglial differences, we first examined expression levels of several microglia-produced cytokines known to be elevated following cuprizone exposure,including tumor necrosis factor alpha (TNFα), interleukin 6(IL-6), interleukin 1 alpha (IL-1α), and transforming growth factor beta (TGFβ) (Arnett et al., 2003; Olah et al., 2012). Although there were some differences in expression at isolated recovery time points between WT and GALC+/–animals, we generally saw similar increases in expression early in recovery (0-, 1-week) that declined at later recovery time points.
Examination of the microglial relationship with myelin debris during the recovery period, however, revealed striking differences in uptake of this debris in GALC+/–mice. Clearance of myelin debris is a process in which microglia play a critical role (Napoli and Neumann, 2010; Olah et al., 2012;Voss et al., 2012; Gudi et al., 2014; Lampron et al., 2015).When we analyzed the number of microglia co-localized with myelin (Iba-1/FluoroMyelin double-positive cells), we found fewer double positive microglia at 2- and 3-week post-cuprizone treatment in the GALC+/–animals compared to WT mice. This outcome suggests the GALC+/–microglia were less effective at myelin phagocytosis compared to WT microglia.
It was particularly intriguing that decreased clearance of myelin debris in the CNS of GALC+/–mice only became apparent as recovery was progressing. It has been shown recently that microglia can become overburdened when processing myelin debris, resulting in lysosomal inclusions and neuroimmunological dysfunction (Safaiyan et al., 2016). One possible interpretation of our observations is that GALC+/–microglia may be hypersensitive to overload, resulting in lower thresholds of myelin debris that can be efficiently phagocytosed and eliminated. Why this defect was not apparent in our studies during the period of cuprizone treatment itself is not known,but it may be that cuprizone modifies microglial function such that other aspects of function of these cells are dominant during the actual time of treatment.
GALC+/– microglia show compromised elevation of proteins important in uptake of damaged myelin
Another indication that GALC+/–microglia were defective in their response to myelin debris was that microglia from heterozygous mutant mice showed decreased elevation of proteins critical in this response. Microglia from GALC+/–mice had decreased expression of Triggering receptor expressed on myeloid cells-2 (Trem2), which is thought to play such an important role in microglial clearance of myelin debris that elevated expression of Trem2 has been used as a readout of myelin phagocytosis after cuprizone exposure (Cantoni et al., 2015; Lampron et al., 2015; Poliani et al., 2015). Trem2 in cuprizone-injured animals was signi ficantly decreased in GALC+/–animals when compared to WT recovery-matched animals at the same time points when there also were significant decreases in numbers of Iba1/FluoroMyelin™ positive microglia (i.e., 2 and 3 weeks after cuprizone was removed from the diet). Trem2 was not the only microglial protein that differed in its expression, however, and we also found a signi ficant reduction in expression of alphaM/beta2 integrin complement receptor-3 (MAC-1/CR3), another microglial receptor involved in myelin debris phagocytosis (Rotshenker, 2003), at 2 weeks of recovery.
In vitro studies on isolated microglia demonstrated that harboring a heterozygous loss-of-function at the GALC locus alters the ability of these cells to engulf myelin debris and to increase Trem2 levels when exposed to such debris (as summarized inFigure 2). In these experiments, microglia and myelin debris were isolated from brains of 3-month-old WT and GALC+/–littermates (n = 3 animals per genotype;Lee and Tansey, 2013). The isolated myelin debris was labeled with the lipophillic dye Di-I, and cells were exposed to labeled myelin debris overnight. We observed that phagocytic-mediated endocytosis was impaired and significantly less myelin was engulfed by GALC+/–cells than by WT microglia. The GALC+/–microglia also showed a decreased elevation of Trem2 expression compared to WT cells when challenged with myelin debris. In WT microglia exposed to myelin debris, there was a nearly six-fold increase in Trem2 expression. In contrast, in vitro exposure to myelin debris did not cause an increase in Trem-2 expression in microglia isolated from GALC+/–animals.
Myelin phagocytosis and Trem2 expression are enhanced in GALC+/– microglia in vitro by NKH-477, a drug that corrects lysosomal dysfunction in models of KD in vitro and in vivo
While we do not know what causes the failure of myelin debris uptake in GALC+/–microglia, our previous studies showed that GALC–/–O2A/OPCs, or WT O-2A/OPCs exposed to psychosine, show multiple lysosomal-related defects. These defects include a failure of endolysosomal processing as measured by uptake of fluorescently labeled dextran beads (Folts et al., 2016).
In our previous studies on KD and other LSDs we also identified several drugs with the unexpected property of restoring normal lysosomal and cellular function in cells exposed to psychosine. We further showed that one of these drugs, NKH-477 (a.k.a. colforsin, a water-soluble derivative of forskolin that is approved for treatment of acute heart failure in Japan (No authors listed, 1999)) provided multiple benefits in vivo (including rescue of myelin damage and extension of lifespan) in twi GALC–/–mice. NKH-477 treatment also restored normal endolysosomal trafficking,lysosomal pH and processing of neutral triglycerides and phospholipids in WT O2A/OPCs exposed to psychosine in vitro (Folts et al., 2016)
When we exposed GALC+/–microglia to myelin debris together with NKH-477, we found that this drug increased myelin phagocytosis and Trem2 expression in the GALC+/–cells. Thus, one of the protective agents discovered in our studies on KD was able, at least in vitro, to overcome the defects in the GALC+/–microglia. Interestingly, this drug did not alter myelin phagocytosis or Trem2 expression in WT microglia exposed to myelin debris, and effects seemed limited to restoring normal function in defective cells rather than enhancing function generally in both GALC+/–and WT cells.
Implications of our findings
Does defective repair provide a new perspective on disease vulnerability?
The finding that being a heterozygous carrier of a disease-causing mutation can compromise the repair of tissue damage appears to provide a new mechanism by which vulnerability to disease could be increased. It is recognized that genetic variants, and/or environmental factors that increase the risk of disease, can cause damage directly and/or can increase the risk that damage will occur. In addition, contributions to disease pathogenesis characterized by a failure to repair damage are recognized to occur at the molecular level; for example, compromised repair of DNA lesions is seen in individuals with diseases, such as Fanconi’s anemia,in which DNA repair enzyme function is altered by mutation (e.g., van der Heijden et al., 2003; Vrouwe et al., 2007).At the cellular level, however, our studies appear to offer the first example of a vulnerability factor, and of a recessive genetic disease, in which heterozygotes show speci fic defects in the ability to repair tissue damage.
It seems unlikely that compromising the ability to repair lesions is unique to mutations in GALC. For example, recent studies reported that migration of O2A/OPCs in the CNS is inhibited in cells expressing the U94A latency-associated protein of human herpesvirus-6 (HHV6) (Campbell et al.,2017), and it is thought that insufficient migration also can contribute to poor repair of myelin damage (Boyd et al.,2013). HHV6 infections are endemic in the human population, and it is likely that most individuals harbor a latent HHV6 infection in their CNS. More importantly, HHV6 appears to be unique in its ability to integrate into human chromosomes and thus become inherited via germline transmission (Arbuckle and Medveczky, 2011; Kuhl et al.,2015). Such integration is not uncommon, and may affect about 1% of the population (Clark, 2016; Pantry and Medveczky, 2017).
Possible relevance of our findings to MS
The defects in myelin debris clearance and microglial function caused by GALC mutation offer an alteration in biological function that is attractive for its potential relevance to MS. Repair of myelin damage grows increasingly limited as MS progresses, leading to severe neurological disability. For multiple reasons, it has even been suggested that the inability to efficiently repair demyelinated lesions is what ultimately leads to the development of progressive MS (Kuhlmann,2008; Voss et al., 2012; Dutta and Trapp, 2014; Harlow et al.,2015; Mahad et al., 2015; Kipp, 2016; Ontaneda et al., 2017).The lack of repair would itself compromise neuronal function, and the unprocessed debris would additionally provide a continued reservoir of damaged myelin that would serve as an endogenous vaccine that would expand the range of the auto-immune attack against white matter through the process of epitope spreading.
In addition, the presence of damaged myelin also can prevent the differentiation of O2A/OPCs into oligodendrocytes, which would limit the number of cells responsible for the production of new myelin. This failure of repair, with associated neuronal dysfunction, would also contribute to irreversible degeneration (Kotter et al., 2006).
Alterations in function of microglia, the CNS-resident immune cells, also could be important in multiple aspects of MS pathology (Napoli and Neumann, 2010; Voss et al.,2012; Gudi et al., 2014; Harlow et al., 2015; Lampron et al.,2015). These cells contribute to the neuroin flammation that can cause widespread tissue damage, and also play critical roles in normal neural development, tissue surveillance and repair, and in the clearance of debris that is necessary for repair (Matsushima and Morell, 2001; Napoli and Neumann,2010; Paolicelli et al., 2011; Franklin et al., 2012; Olah et al.,2012; Voss et al., 2012; Gudi et al., 2014; Harlow et al., 2015;Lampron et al., 2015; Hong et al., 2016; Kipp, 2016).
Defects in Trem2 elevation could be relevant to MS
The failure of GALC+/–microglia to elevate levels of Trem2 in response to myelin debris is a finding of particular interest for multiple reasons. Trem2 appears to be critical for the response to demyelination (Cantoni et al., 2015; Poliani et al., 2015), and loss of Trem2 function leads to impaired remyelination following cuprizone-induced demyelination,much like the phenotype we observed in GALC+/–mice. Engagement of Trem2 decreases inflammation, and blockade of Trem2 worsens disease progress in the experimental allergic encephalomyelitis (EAE) animal model of MS (Piccio et al., 2007). Conversely, transplantation of myeloid precursor cells genetically modi fied to express Trem2 limits tissue destruction in this same model (Takahashi et al., 2007). In humans, loss-of-function mutations in Trem2 cause the rare Nasu-Hakola disease, which is associated with demyelination of subcortical white matter damage and lethal early onset dementia (Neumann and Takahashi, 2007). Thus, a failure in Trem2 elevation may contribute to several different outcomes relevant to the MS phenotype.
Defects in Trem2 function also may have implications that extend far beyond illnesses with myelin damage as a critical contribution to disease pathology, and that will warrant attention in further studies on the effects of the heterozygous mutations in the GALC gene. Alterations in Trem2 function are of active interest in respect to Alzheimer’s disease and potentially other neurodegenerative disorders (Jiang et al.,2013, 2014; Rohn, 2013; Jay et al., 2015; Lill et al., 2015; Lue et al., 2015; Painter et al., 2015; Poliani et al., 2015; Wang et al., 2015, 2016; Walter, 2016; Han et al., 2017). For example,in respect to microglial function, Trem2 is also critical for clearance of neuronal apoptosis (Takahashi et al., 2005) and of amyloid plaques in the presence of anti-amyloid beta antibodies (Xiang et al., 2016). Trem2 knockdown also worsens outcomes in experimental stroke models (Kawabori et al.,2015) and Trem2 loss may even generally accelerate aging processes (Poliani et al., 2015; Raha et al., 2017). Thus, the relevance of a defect in mobilization of Trem2 could have relevance to the pathology of a broad range of diseases.
It also is of interest to speculate whether our findings on microglial dysfunction are relevant to KD itself. Could failures in normal microglial function themselves contribute to the massive white matter damage in this disease?
Understanding the biology of vulnerability may lead to new treatments for neurodegenerative disorders

Figure 2 Summary of differences between unmanipulated twitcher (twi) heterozygotes (GALC+/–) microglia and wild-type (WT) microglia exposed to myelin debris in vitro.
One of the intriguing examples of how studies on genetic contributions to disease vulnerability may have therapeutic implications is seen in studies on the relationship between GD and Parkinson’s disease. Based on findings that 5–10%of Parkinson’s patients may have mutations in glucocerebrosidase (the enzyme mutated in GD), there is an emerging interest in using molecular chaperones to stabilize and increase the activity of this enzyme in Parkinson’s patients harboring such mutations with the hope of slowing disease progression (e.g., Ambrosi et al., 2015; Migdalska-Richards et al., 2016, 2017; Ishay et al., 2018).
Analogously to studies on GD and PD, one of the most encouraging outcomes of our own experiments was the demonstration that it is possible to enhance myelin debris uptake and Trem2 expression in GALC+/–microglia using one of the drugs discovered in our research on KD and other LSDs (Folts et al., 2016). Impaired myelin debris clearance is thought to contribute to the inefficient remyelination that is often observed in MS patients, particularly in patients with progressive MS where the imbalance of demyelination and remyelination leads to severe neurological disability. Currently, there are no efficacious treatment options for progressive MS patients (Dutta and Trapp, 2014; Ontaneda et al., 2017).Thus, there exists a great need to better identify underlying disease mechanisms, particularly those that contribute to the impaired remyelination associated with the transition into progressive MS, and to identify better therapeutic options for these patients. While much further work needs to be done to determine whether the approach we are studying might be of therapeutic relevance in MS, it is nonetheless a promising first step in demonstrating that it is possible to identify pharmacological agents that normalize myelin debris uptake even in mutationally altered GALC+/–microglia.
Additional next steps
One of the central questions to emerge from the study of effects of heterozygosity for mutations that cause KD or GD is the extent to which such findings will apply to other LSDs.For example, we recently showed that there may be general principles relevant to understanding and preventing the cellular and lysosomal abnormalities that occur in at least four of the spingolipidoses (a sub-family of LSDs that includes KD, GD, metachromatic leukodystrophy, Fabry disease,Tay-Sachs disease, Niemann-Pick disease). It will be of great interest to determine if our findings on changes in lesion repair and/or microglial function also occur in heterozygous carriers of mutations that cause other LSDs.
Two lines of investigation would greatly speed progress in answering the above question. The first of these would be for the foundations that are interested in LSDs to obtain detailed family histories that might reveal increases in the occurrence of speci fic other diseases. Such information may provide use-ful clues on the types of pathological changes to study.
In addition, a targeted sequencing of genes involved in LSDs seems like it would be warranted, at a minimum in populations for which these genes are established as risk loci or that have pathologies that resemble an LSD in at least some features. For example, it may be of future interest to directly determine the prevalence of GALC mutations in MS patients. There has been a single report of a child with exceptionally severe KD born into a family in which several members had MS (Sahai et al., 2005). It also is intriguing that analysis of 56 MS patients for mutations causing pseudodeficiency in arylsulfatase-A (the mutation of which causes the demyelinating LSD of metachromatic leukodystrophy) revealed mutations in 13 of these patients. Moreover, patients with mutations had a greater total number of lesions and the number of hypo-intense lesions on T1-weighted images was greater in MS patients carrying such mutations (Baronica et al., 2011).
The potential relevance of GALC mutations and arylsulfatase-A mutations to MS also raises the question of whether therapies targeting the biological consequences of such mutations might be of relevance in MS treatment, as suggested by the outcome of our studies on NKH-477. While much further work needs to be done to determine whether this approach might be of therapeutic relevance in MS, it is nonetheless a promising first step in demonstrating that it is possible to identify pharmacological agents that normalize myelin debris uptake even in mutationally altered GALC+/–microglia. In addition, if GALC mutations are as common in MS patients as suggested for Arylsulfatase-A mutations, then there might also be therapeutic utility in treatments aimed at improving GALC enzymatic activity in MS patients using approaches being explored as possible KD therapies (Lee et al., 2010; Berardi et al., 2014; Hill et al., 2015; Hossain et al.,2015; Graziano et al., 2016; Spratley et al., 2016).
Increasing evidence is linking genes known to cause LSDs with neurodegenerative diseases. Although we do not yet understand the functional roles of many of these associations, it is possible that changes in underlying lysosomal biology could contribute to the development of disease symptoms as well as the failure of repair mechanisms that often occurs concurrently. Thus, treatments identified for LSD patients may also be able to provide therapeutic bene fits for patients with neurodegenerative diseases, a possibility that warrants further exploration.
Author contributions:All authors contributed to the writing of this manuscript. The experiments discussed were conducted by NJSH and CJF.
Con flicts of interest:The authors have declared that no con flicts of interest exist.
Financial support:This research we conducted on twitcher GALC+/–heterozygotes was supported by the following funding sources: National Institutes of Health, F31-NS078911, https://www.nih.gov (NSH); New York State Department of Health, NYS-DOH-C026877, http://www.stemcell.ny.gov (NSH); New York State Department of Health, NYSDOH-C029557, http://www.stemcell.ny.gov (MN); New York State Department of Health, NYS-DOH-C026877, http://www.stemcell.ny.gov(CJF); Hunter’s Hope, http://www.huntershope.org/site/PageServer(MN); Children’s Neurobiological Solutions Foundation, http://pediatricbrainfoundation.org (MN); and the Legacy of Angels, http://tloaf.org (MN).
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under identical terms.
Open peer review report:
Reviewer:Una FitzGerald, National University of Ireland - Galway,Ireland.
Comments to authors:This review summarized findings relating to heterozygous carriers of mutations in galactocerebrosidase (GALC)and their possible relevance to multiple sclerosis (MS). A discussion of Krabbe and Gaucher disease, two lysosomal storage disorders, leads to more detailed exposition of the effects of GALC mutations on the repair of cuprizone-induced demyelination and factors that mediate a defective microglial response to same. The review finishes by proposing that manipulation of triggering receptor expressed in myeloid cells-2 (TREM-2)and other molecules associated with myelin debris clearance by microglia, could represent a novel and productive approach to boosting remyelination and restoration of function in MS. The utility of tracking GALC mutation incidence amongst the MS community is also suggested as an important contributor to the understanding of the molecular mechanisms underlying MS disease incidence and course within different individuals. The authors have provided an engaging and highly informative summary of the possible importance of galactocerebrosidase function in MS incidence and disease. In what is a very well-written manuscript,the authors take the reader logically from LSDs to the speci fics of GALC variants and their suggested impact on the repair of demyelination.Sub-headings such as ‘Heterozygous loss of GALC function causes and impaired microglial response to myelin damage’ provide informative summaries of section content and entice further enthusiastic reading.This reviewer very much enjoyed reading the review and learned some interesting important information about CNS myelin repair.
Ambrosi G, Ghezzi C, Zangaglia R, Levandis G, Pacchetti C, Blandini F (2015) Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol Dis 82:235-242.
Arbuckle JH, Medveczky PG (2011) The molecular biology of human herpesvirus-6 latency and telomere integration. Microbes Infect 13:731-741.
Arnett HA, Wang Y, Matsushima GK, Suzuki K, Ting JP (2003) Functional genomic analysis of remyelination reveals importance of in flammation in oligodendrocyte regeneration. J Neurosci 23:9824-9832.
Barcellos LF, Sawcer S, Ramsay PP, Baranzini SE, Thomson G, Briggs F, Cree BC, Begovich AB, Villoslada P, Montalban X, Uccelli A,Savettieri G, Lincoln RR, DeLoa C, Haines JL, Pericak-Vance MA,Compston A, Hauser SL, Oksenberg JR (2006) Heterogeneity at the HLA-DRB1 locus and risk for multiple sclerosis. Hum Mol Genet 15:2813-2824.
Baronica KB, Mlinac K, Ozretic D, Vladic A, Bognar SK (2011) Arylsulfatase a gene polymorphisms in relapsing remitting multiple sclerosis: genotype-phenotype correlation and estimation of disease progression. Coll Antropol 35 Suppl 1:11-16.
Bashinskaya VV, Kulakova OG, Boyko AN, Favorov AV, Favorova OO(2015) A review of genome-wide association studies for multiple sclerosis: classical and hypothesis-driven approaches. Hum Genet 134:1143-1162.
Berardi AS, Pannuzzo G, Graziano A, Costantino-Ceccarini E, Piomboni P, Luddi A (2014) Pharmacological chaperones increase residual beta-galactocerebrosidase activity in fibroblasts from Krabbe patients. Mol Genet Metab 112:294-301.
Boyd A, Zhang H, Williams A (2013) Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol 125:841-859.
Campbell A, Hogestyn JM, Folts CJ, Lopez B, Proschel C, Mock D,Mayer-Proschel M (2017) Expression of the human herpesvirus 6A latency-associated transcript U94A disrupts human oligodendrocyte progenitor migration. Sci Rep 7:3978.
Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R,Yuede CM, Galimberti D, Olivecrona G, Klein RS, Cross AH, Otero K, Piccio L (2015) TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol 129:429-447.
Clark DA (2016) Clinical and laboratory features of human herpesvirus 6 chromosomal integration. Clin Microbiol Infect 22:333-339.
Dutta R, Trapp BD (2014) Relapsing and progressive forms of multiple sclerosis: insights from pathology. Curr Opin Neurol 27:271-278.
Folts CJ, Scott-Hewitt N, Pröschel C, Mayer-Pröschel M, Noble M(2016) Lysosomal re-acidi fication prevents lysosphingolipid-induced lysosomal impairment and cellular toxicity. PLoS Biol 14:e1002583.
Franklin RJ, ffrench-Constant C, Edgar JM, Smith KJ (2012) Neuroprotection and repair in multiple sclerosis. Nat Rev Neurol 8:624-634.
George MF, Briggs FB, Shao X, Gianfrancesco MA, Kockum I, Harbo HF, Celius EG, Bos SD, Hedstrom A, Shen L, Bernstein A, Alfredsson L, Hillert J, Olsson T, Patsopoulos NA, De Jager PL, Oturai AB,Sondergaard HB, Sellebjerg F, Sorensen PS, et al. (2016) Multiple sclerosis risk loci and disease severity in 7,125 individuals from 10 studies. Neurol Genet 2:e87.
Graziano AC, Pannuzzo G, Avola R, Cardile V (2016) Chaperones as potential therapeutics for Krabbe disease. J Neurosci Res 94:1220-1230.
Gudi V, Gingele S, Skripuletz T, Stangel M (2014) Glial response during cuprizone-induced de- and remyelination in the CNS: lessons learned. Front Cell Neurosci 8:73.
Han J, Wang M, Ren M, Lou H (2017) Contributions of triggering-receptor-expressed-on-myeloid-cells-2 to neurological diseases. Int J Neurosci 127:368-375.
Harlow DE, Honce JM, Miravalle AA (2015) Remyelination therapy in multiple sclerosis. Front Neurol 6:257.
Hill CH, Viuff AH, Spratley SJ, Salamone S, Christensen SH, Read RJ,Moriarty NW, Jensen HH, Deane JE (2015) Azasugar inhibitors as pharmacological chaperones for Krabbe disease. Chem Sci 6:3075-3086.
Hong S, Dissing-Olesen L, Stevens B (2016) New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol 36:128-134.
Hossain MA, Higaki K, Saito S, Ohno K, Sakuraba H, Nanba E, Suzuki Y, Ozono K, Sakai N (2015) Chaperone therapy for Krabbe disease:potential for late-onset GALC mutations. J Hum Genet 60:539-545.
Ihara M, Polvikoski TM, Hall R, Slade JY, Perry RH, Oakley AE, Englund E, O’Brien JT, Ince PG, Kalaria RN (2010) Quanti fication of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer’s disease, and dementia with Lewy bodies. Acta Neuropathol 119:579-589.
Ishay Y, Zimran A, Szer J, Dinur T, Ilan Y, Arkadir D (2018) Combined beta-glucosylceramide and ambroxol hydrochloride in patients with Gaucher related Parkinson disease: From clinical observations to drug development. Blood Cells Mol Dis 68:117-120
Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998)Microglia-specific localisation of a novel calcium binding protein,Iba1. Brain Res Mol Brain Res 57:1-9.
Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML,Xu G, Margevicius D, Karlo JC, Sousa GL, Cotleur AC, Butovsky O, Bekris L, Staugaitis SM, Leverenz JB, Pimplikar SW, Landreth GE, Howell GR, Ransohoff RM, Lamb BT (2015) TREM2 de ficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med 212:287-295.
Jiang T, Yu JT, Zhu XC, Tan L (2013) TREM2 in Alzheimer’s disease.Mol Neurobiol 48:180-185.
Jiang T, Tan L, Zhu XC, Zhang QQ, Cao L, Tan MS, Gu LZ, Wang HF,Ding ZZ, Zhang YD, Yu JT (2014) Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 39:2949-2962.
Jokubaitis VG, Butzkueven H (2016) A genetic basis for multiple sclerosis severity: Red herring or real? Mol Cell Probes 30:357-365.
Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL,Nakamura MC, Yenari MA (2015) Triggering receptor expressed on myeloid cells 2 (TREM2) de ficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci 35:3384-3396.
Kipp M (2016) Remyelination strategies in multiple sclerosis: a critical re flection. Expert Rev Neurother 16:1-3.
Kipp M, Clarner T, Dang J, Copray S, Beyer C (2009) The cuprizone animal model: new insights into an old story. Acta Neuropathol 118:723-736.
Kotter MR, Li WW, Zhao C, Franklin RJM (2006) Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci 26:328-332.
Kuhl U, Lassner D, Wallaschek N, Gross UM, Krueger GR, Seeberg B,Kaufer BB, Escher F, Poller W, Schultheiss HP (2015) Chromosomally integrated human herpesvirus 6 in heart failure: prevalence and treatment. Eur J Heart Fail 17:9-19.
Kuhlmann T (2008) Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131:1749-1758.
Kukita Y, Furuya H, Kobayashi T, Sakai N, Hayashi K (1997) Characterization of the GALC gene in three Japanese patients with adult-onset Krabbe disease. Genet Test 1:217-223.
Lampron A, Larochelle A, Laflamme N, Prefontaine P, Plante MM,Sanchez MG, Yong VW, Stys PK, Tremblay ME, Rivest S (2015)Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med 212:481-495.
Lee JH, Cho MH, Hersh CP, McDonald ML, Wells JM, Drans field MT,Bowler RP, Lynch DA, Lomas DA, Crapo JD, Silverman EK, COPDGene and ECLIPSE Investigators (2015) IREB2 and GALC are associated with pulmonary artery enlargement in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 52:365-376.
Lee JK, Tansey MG (2013) Microglia isolation from adult mouse brain.Methods Mol Biol 1041:17-23.
Lee WC, Kang D, Causevic E, Herdt AR, Eckman EA, Eckman CB(2010) Molecular characterization of mutations that cause globoid cell leukodystrophy and pharmacological rescue using small molecule chemical chaperones. J Neurosci 30:5489-5497.
Lill CM, Rengmark A, Pihlstrom L, Fogh I, Shatunov A, Sleiman PM,Wang LS, Liu T, Lassen CF, Meissner E, Alexopoulos P, Calvo A,Chio A, Dizdar N, Faltraco F, Forsgren L, Kirchheiner J, Kurz A,Larsen JP, Liebsch M, et al. (2015) The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration,amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimers Dement 11:1407-1416.
Liu Y, Gibson J, Wheeler J, Kwee LC, Santiago-Turla CM, Akafo SK,Lichter PR, Gaasterland DE, Moroi SE, Challa P, Herndon LW,Girkin CA, Budenz DL, Richards JE, Allingham RR, Hauser MA(2011) GALC deletions increase the risk of primary open-angle glaucoma: the role of Mendelian variants in complex disease. PLoS One 6:e27134.
Lue LF, Schmitz C, Walker DG (2015) What happens to microglial TREM2 in Alzheimer’s disease: Immunoregulatory turned into immunopathogenic? Neuroscience 302:138-150.
Mahad DH, Trapp BD, Lassmann H (2015) Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 14:183-193.
Marshall MS, Bongarzone ER (2016) Beyond Krabbe’s disease: The potential contribution of galactosylceramidase deficiency to neuronal vulnerability in late-onset synucleinopathies. J Neurosci Res 94:1328-1332.
Matsuo A, Lee GC, Terai K, Takami K, Hickey WF, McGeer EG,McGeer PL (1997) Unmasking of an unusual myelin basic protein epitope during the process of myelin degeneration in humans: a potential mechanism for the generation of autoantigens. Am J Pathol 150:1253-1266.
Matsushima GK, Morell P (2001) The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol 11:107-116.
Migdalska-Richards A, Daly L, Bezard E, Schapira AH (2016) Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice.Ann Neurol 80:766-775.
Migdalska-Richards A, Ko WKD, Li Q, Bezard E, Schapira AHV (2017)Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse 71.
Napoli I, Neumann H (2010) Protective effects of microglia in multiple sclerosis. Exp Neurol 225:24-28.
Neumann H, Takahashi K (2007) Essential role of the microglial triggering receptor expressed on myeloid cells-2 (TREM2) for central nervous tissue immune homeostasis. J Neuroimmunol 184:92-99.
Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH,Li A, Holton J, Guerreiro R, Paudel R, Segarane B, Singleton A, Lees A, Hardy J, Houlden H, Revesz T, Wood NW (2009) Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132:1783-1794.
No authors listed (1999) In: WHO Pharmaceutical Newsletter No. 9 &12: World Health Organization, Geneva, Switzerland.
O’Regan G, deSouza RM, Balestrino R, Schapira AH (2017) Glucocerebrosidase Mutations in Parkinson Disease. J Parkinsons Dis 7:411-422.
Olah M, Amor S, Brouwer N, Vinet J, Eggen B, Biber K, Boddeke HW(2012) Identi fication of a microglia phenotype supportive of remyelination. Glia 60:306-321.
Ontaneda D, Thompson AJ, Fox RJ, Cohen JA (2017) Progressive multiple sclerosis: prospects for disease therapy, repair, and restoration of function. Lancet 389:1357-1366.
Painter MM, Atagi Y, Liu CC, Rademakers R, Xu H, Fryer JD, Bu G(2015) TREM2 in CNS homeostasis and neurodegenerative disease.Mol Neurodegener 10:43.
Pantry SN, Medveczky PG (2017) Latency, integration, and reactivation of human herpesvirus-6. Viruses 9.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P,Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456-1458.
Piccio L, Buonsanti C, Mariani M, Cella M, Gil fillan S, Cross AH, Colonna M, Panina-Bordignon P (2007) Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol 37:1290-1301.
Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gil fillan S, Colonna M (2015) TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest 125:2161-2170.
Praet J, Guglielmetti C, Berneman Z, Van der Linden A, Ponsaerts P (2014) Cellular and molecular neuropathology of the cuprizone mouse model: clinical relevance for multiple sclerosis. Neurosci Biobehav Rev 47:485-505.
Raha AA, Henderson JW, Stott SR, Vuono R, Foscarin S, Friedland RP,Zaman SH, Raha-Chowdhury R (2017) Neuroprotective Effect of TREM-2 in Aging and Alzheimer’s Disease Model. J Alzheimers Dis 55:199-217.
Rohn TT (2013) The triggering receptor expressed on myeloid cells 2:“TREM-ming” the in flammatory component associated with Alzheimer’s disease. Oxid Med Cell Longev 2013:860959.
Rotshenker S (2003) Microglia and macrophage activation and the regulation of complement-receptor-3 (CR3/MAC-1)-mediated myelin phagocytosis in injury and disease. J Mol Neurosci 21:65-72.
Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S,Edinger AL, Jung S, Rossner MJ, Simons M (2016) Age-related myelin degradation burdens the clearance function of microglia during aging. Nat Neurosci 19:995-998.
Sahai I, Baris H, Kimonis V, Levy HL (2005) Krabbe disease: severe neonatal presentation with a family history of multiple sclerosis. J Child Neurol 20:826-828.
Sawcer S, Franklin RJ, Ban M (2014) Multiple sclerosis genetics. Lancet Neurol 13:700-709.
Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA,Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, Edkins S, Gray E, Booth DR, Potter SC, Goris A, Band G, Oturai AB, Strange A,Saarela J, Bellenguez C, et al. (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476:214-219.
Schapira AH (2015) Glucocerebrosidase and Parkinson disease: Recent advances. Mol Cell Neurosci 66:37-42.
Spratley SJ, Hill CH, Viuff AH, Edgar JR, Skjodt K, Deane JE (2016)Molecular mechanisms of disease pathogenesis differ in Krabbe disease variants. Traffic 17:908-922.
Suzuki K (2003) Globoid cell leukodystrophy (Krabbe’s disease): update. J Child Neurol 18:595-603.
Takahashi K, Rochford CD, Neumann H (2005) Clearance of apoptotic neurons without in flammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 201:647-657.
Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H (2007)TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med 4:e124.
Tappino B, Biancheri R, Mort M, Regis S, Corsolini F, Rossi A, Stroppiano M, Lualdi S, Fiumara A, Bembi B, Di Rocco M, Cooper DN, Filocamo M (2010) Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease. Hum Mutat 31:E1894-1914.
van der Heijden MS, Yeo CJ, Hruban RH, Kern SE (2003) Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res 63:2585-2588.
Voss EV, Skuljec J, Gudi V, Skripuletz T, Pul R, Trebst C, Stangel M(2012) Characterisation of microglia during de- and remyelination:can they create a repair promoting environment? Neurobiol Dis 45:519-528.
Vrouwe MG, Elghalbzouri-Maghrani E, Meijers M, Schouten P, Godthelp BC, Bhuiyan ZA, Redeker EJ, Mannens MM, Mullenders LH,Pastink A, Darroudi F (2007) Increased DNA damage sensitivity of Cornelia de Lange syndrome cells: evidence for impaired recombinational repair. Hum Mol Genet 16:1478-1487.
Walter J (2016) The triggering receptor expressed on myeloid cells 2: a molecular link of neuroin flammation and neurodegenerative diseases. J Biol Chem 291:4334-4341.
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML,Gil fillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, Holtzman DM, Cirrito JR, Colonna M (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160:1061-1071.
Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gil fillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, Colonna M (2016) TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 213:667-675.
Wenger DA (1993) Research update on lysosomal disorders with special emphasis on metachromatic leukodystrophy and Krabbe disease.APMIS Suppl 40:81-87.
Wenger DA, Sattler M, Hiatt W (1974) Globoid cell leukodystrophy:deficiency of lactosyl ceramide beta-galactosidase. Proc Natl Acad Sci U S A 71:854-857.
Wenger DA, RafiMA, Luzi P (1997) Molecular genetics of Krabbe disease (globoid cell leukodystrophy): diagnostic and clinical implications. Hum Mutat 10:268-279.
Wenger DA, Ra fiMA, Luzi P, Datto J, Costantino-Ceccarini E (2000)Krabbe disease: genetic aspects and progress toward therapy. Mol Genet Metab 70:1-9.
Xiang X, Werner G, Bohrmann B, Liesz A, Mazaheri F, Capell A,Feederle R, Knuesel I, Kleinberger G, Haass C (2016) TREM2 de ficiency reduces the efficacy of immunotherapeutic amyloid clearance.EMBO Mol Med 8:992-1004.
- 中国神经再生研究(英文版)的其它文章
- The biological clock: future of neurological disorders therapy
- Cerebral ischemia and neuroregeneration
- SNARE complex in axonal guidance and neuroregeneration
- The relaxin peptide family – potential future hope for neuroprotective therapy? A short review
- Roles of neural regeneration in memory pharmacology
- Towards frequency adaptation for delayed feedback deep brain stimulations