
基质固相分散萃取和固相萃取-高效液相色谱-三重四极杆-复合线性离子阱质谱同时测定奶制品中6种雌激素
2018-04-02 06:50:23韩疏影俞慧敏宋易霖邓海山池玉梅南京中医药大学药学院江苏南京210023
色谱 2018年3期
韩疏影, 俞慧敏, 宋易霖, 邓海山, 柴 川, 池玉梅(南京中医药大学药学院, 江苏 南京 210023)
外源性雌激素(如雌三醇(E3)、雌酮(E1)、17β-雌二醇(17β-E2)、17α-雌二醇(17α-E2)、炔雌醇(EE)及孕酮(P4)等)及其衍生物是常见的内分泌干扰物。研究发现,癌症(如睾丸癌、前列腺癌、乳腺癌和卵巢癌等)和生殖疾病(如男性精液质量差等)都可能与体内雌激素表达水平增加有关[1-6]。1981年世界卫生组织召开的“雌激素对胎儿发育和婴儿健康”会议中就针对外源性雌激素在婴儿体内半衰期较长、摄入过量是否会对机体产生危害进行了讨论。目前,外源性雌激素对于人体的潜在危害已引起了广泛关注[7,8]。
雌激素常被非法用作奶牛等动物的生长促进剂,以加快动物的生长、增加动物的体重,从而导致牛奶中雌激素含量超标[9]。欧盟自1988年起禁止将甾体激素用于促生长,并对动物源食品制定了系列监管措施[10]。2002年我国明确立法禁止性激素和具有雌激素样作用的物质在食品动物中使用,外源性激素物质在动物性食品中不得检出[11]。因此,对于复杂基质中雌激素残留的监测,需要建立灵敏度高、准确性好、简便易行的分析方法。
目前,生物样品中类固醇类激素样物质的分析检测方法主要有液相色谱-紫外检测法(LC-UV)[12]、液相色谱-荧光检测法(LC-FLD)[13]、液相色谱-质谱联用法(LC-MS)[14,15]、气相色谱-质谱联用法(GC-MS)[16]、薄层色谱法(TLC)[17]和高效薄层色谱法(HPTLC)[18]等。其中,LC-MS以其良好的定性和定量性能,逐渐成为检测类固醇类雌激素的主要方法,但其精密度和准确度易受基质效应的影响。动物源食品基质复杂,激素和激素样物质种类多、水平低(μg/kg~ng/kg),必须配以合适的预处理技术进行富集提取,并用特异性高的方法实现多种结构类似物的分离。
目前,已有多篇报道将固相微萃取(SPME)、液相微萃取(LPME)等为代表的微处理技术应用于激素样品的预处理中[19,20]。微处理技术的重复性和精密度虽逊于SPE,但其兼具分离和富集功能,且可与液相色谱体系良好兼容[21]。基质固相分散萃取(MSPD)兼具分散、萃取及净化功能,且精密度更高,可用于生物样品中抗生素类、激素及激素样物质的提取和净化[22,23]。目前MSPD在农残测定中已广泛用于固体、半固体及高黏度的基质样品[24],但其用于提取奶制品中的雌激素仅见于液体基质[25,26]。
本文采用MSPD和SPE法分别提取了固体奶粉和液体牛奶中E3、E1、17β-E2、17α-E2、EE及P4等6种雌激素,并采用高效液相色谱-三重四极杆-复合线性离子阱质谱(HPLC-Q-TRAP-MS)技术建立了操作简便、分析快速、试样和有机溶剂用量少、准确性高和重复性好的测定方法,以期为准确定量奶制品中雌激素含量奠定基础。
1 实验部分
1.1 仪器、试剂与材料
ABSciex Q-TRAP 5500质谱仪(ABSciex,美国); LC-20A高效液相色谱仪,配有DGU-20A3在线脱气机、LC-20AD XR泵、SIL-20A XR自动进样器及CTO-20AC柱温箱(Shimadzu,日本); KH-700DE型超声机(昆山禾创超声仪器有限公司); TGL-16G型离心机(上海安亭科学仪器厂)。
甲醇(MeOH)、乙腈(ACN)(均为色谱纯)购自德国Merck公司;乙酸乙酯(EA)、二氯甲烷(DCM)(均为分析纯)购自国药集团化学试剂有限公司;玻璃注射针管购自金坛市第二注射器厂;HLB SPE柱(60 mg/3 mL)、C18(40~60 μm)、硅胶(40~60 μm)和弗罗里硅土(75~100 μm)购自上海安谱实验科技股份有限公司;标准品:E3(纯度97.4%)、E1 (纯度98.3%)、17β-E2 (纯度96.3%)、17α-E2 (纯度97.2%)、EE (纯度98.0%)、P4 (纯度97.5%)和左炔诺孕酮(LNG,纯度99.4%,内标)购自中国药品生物制品检定所。6种雌激素的正辛醇-水分配系数(Kow)及其化学结构分别见表1和图1。实验所用超纯水经Milli-Q系统(美国Millipore公司)净化。
1.2 标准储备液的配制
精密称取6种雌激素各10 mg,分别置于10 mL容量瓶中,用甲醇溶解并定容为1.0 g/L的混合标准溶液;精密称取内标10 mg,置于10 mL容量瓶中,用甲醇溶解并定容为1.0 g/L的内标储备液;混合标准工作溶液(200~0.1 mg/L)由混合标准溶液(1.0 g/L)经30%(v/v)乙腈水溶液逐级稀释得到。

表 1 6种雌激素的Kow值Table 1 Kow data of the six estrogens
Kow: the partition coefficient of solute between water and octanol. LogKowof each compound was calculated by ACD/Lab software.

图 1 6种雌激素的化学结构Fig. 1 Chemical structures of the six estrogens
1.3 样品前处理
1.3.1MSPD
将硅胶吸附剂装入50 mL玻璃注射器至最大刻度,分别用100 mL正己烷、二氯甲烷和甲醇依次洗涤,真空干燥后备用。
精密称取1 g固态奶粉(或量取1 mL液态牛奶,加入无水硫酸镁脱水),置于乳钵中,加入1.0 mL 10 mg/L混合标准工作溶液和10 μL 1.0 g/L内标液,混匀,静置15 min。样品经上述操作后,分别加入预先处理的1 g硅胶吸附剂,研磨均匀,静置30 min后,将其装入底部带有滤纸的10 mL玻璃注射针筒中,轻轻敲打针筒并按压;用10 mL正己烷淋洗,弃去淋洗液,再采用15 mL ACN-EA(4∶1, v/v)洗脱,收集洗脱液,于45 ℃下氮气流吹干,用1 mL 30%(v/v)乙腈水溶液复溶后,以1 500 r/min离心10 min,取5 μL上清液,待HPLC-Q-TRAP-MS分析。
1.3.2SPE
SPE柱使用前依次用6 mL甲醇和6 mL水活化。取1 g固态奶粉,用1 mL水溶解(或1 mL液态牛奶),加入10 μL 1.0 g/L混合标准溶液和10 μL 1.0 g/L内标液,置于10 mL离心管中,加入1 mL甲醇,超声提取,以1 500 r/min离心10 min,将上清液转移至另一10 mL离心管中,加入2 mL乙腈饱和的正己烷萃取,静置分层后弃去正己烷层,氮气流吹干,加入1 mL纯水溶解,上样,用6 mL EA洗脱,收集洗脱液,氮气流吹干,用1 mL 30%(v/v)乙腈水溶液复溶后,以1 500 r/min离心10 min,取上清液5 μL,待HPLC-Q-TRAP-MS分析。
1.4 色谱条件
色谱柱:Cosmosil胆固醇色谱柱(150 mm×4.6 mm, 5 μm,日本Nacalai Tesque公司);柱温:40 ℃;流动相A:水,流动相B:乙腈;流速:0.5 mL/min。梯度洗脱程序:0~5 min, 30%B~40%B; 5~15 min, 40%B; 15~23 min, 40%B~55%B; 23~25 min, 55%B; 25~26 min, 55%B~70%B; 26~31 min, 70%B; 31~32 min, 70%B~30%B。进样量:5 μL;检测波长:220 nm。

表 2 6种雌激素和内标的质谱参数Table 2 MS parameters of the six estrogens and internal standard
DP: declustering potential; CE: collision energy; CXP: collision cell entrance potential; LNG: levonorgestrel.
1.5 质谱条件
离子源:电喷雾离子(ESI)源;采集方式:多反应监测模式(MRM);扫描方式:正负离子同时检测;离子源喷雾电压:5 500 V(正离子模式), -4 500 V(负离子模式);离子源温度:500 ℃;气帘气压力:206.8 kPa;雾化气压力:344.7 kPa;辅助气压力:379.2 kPa。其他质谱条件见表2。
2 结果与讨论
2.1 MSPD与SPE预处理不同奶制品
按1.3节描述,分别采用MSPD和SPE对固态奶粉及液态牛奶进行预处理,固态奶粉及液态牛奶中6种雌激素的回收率结果见表3。结果表明,MSPD法更适合固体奶粉的预处理;SPE法更适合处理液体牛奶。因此后续实验中根据不同的样品选择不同的预处理方法。

表 3 采用基质固相分散萃取和固相萃取处理不同基质时6种雌激素的回收率Table 3 Recoveries of the six estrogens in different sample matrices by matrix solid-phase dispersion (MSPD) and solid-phase extraction (SPE) %
2.2 MSPD与SPE条件的优化
取6种雌激素含量均低于检出限的固态奶粉和液态牛奶作为空白样品,各雌激素对照品的添加量为10 mg/kg(固态奶粉)和10 mg/L(液态牛奶),经HPLC-Q-TRAP-MS分析,比较不同条件下6种雌激素的回收率,从而对MSPD和SPE的条件进行优化。
2.2.1MSPD吸附剂的优化
实验对3种极性差异较大的吸附剂(C18、硅胶和弗罗里硅土)进行考察。参照文献[27]报道,在吸附剂与样品质量比为3∶1、洗脱剂为15 mL中等极性ACN-EA(4∶1, v/v)的条件下对空白奶粉样品进行处理。结果表明,采用C18、硅胶和弗罗里硅土时,6种雌激素的平均回收率分别为66%、71%和56%。C18填料为非极性材料,对非极性和弱极性物质具有较好的溶解性;硅胶则是利用其表面的硅羟基形成分子间氢键而吸附物质;弗罗里硅土是硅酸镁吸附剂,其表面吸附作用强于硅胶。通过图1可知,除P4外,其他5种雌激素均带有羟基,因此当吸附剂的极性越大,对目标物的吸附力也越强,相应洗脱剂的极性也要越大。采用中等极性的洗脱溶剂时,雌激素被吸附于弗罗里硅土中而不能完全被洗脱,因此回收率偏低。6种雌激素的logKow范围为2.94~4.52,疏水性中等,当使用C18填料时,根据相似相溶原理,不能使中等疏水性的雌激素类成分很好地保留。因此,选用硅胶作为MSPD的吸附剂。
2.2.2MSPD吸附剂与样品比例的优化
实验考察了吸附剂与样品的质量比(1∶1、2∶1、3∶1和4∶1)对各雌激素回收率的影响。结果表明,6种雌激素的平均回收率随着吸附剂与样品质量比的增大而逐渐减小,当吸附剂与样品的质量比为1∶1时,6种雌激素的平均回收率为89%,满足分析要求,故选为实验所用。
2.2.3MSPD和SPE洗脱剂的优化
选取从弱极性到极性的溶液DCM、EA、ACN、MeOH和ACN-EA(1∶1、2∶2、3∶1、4∶1、5∶1, v/v),分别采用MSPD和HLB SPE对固态奶粉和液态牛奶进行洗脱,6种雌激素的平均回收率见图2。

图 2 采用(a)MSPD和(b)SPE法时不同洗脱剂对6种雌激素回收率的影响Fig. 2 Effect of different eluents on the recoveries of the six estrogens pretreated by (a) MSPD and (b) SPE DCM: dichloromethane; EA: ethyl acetate; ACN: acetonitrile; MeOH: methanol.
如图2a所示,采用MSPD以硅胶为吸附剂萃取空白奶粉时,极性相对较弱的DCM和EA对6种雌激素的回收率低于67%;逐渐增大洗脱剂的极性,采用MeOH和ACN作为洗脱剂时,回收率明显增加,分别达到75%和72%;采用ACN中加入不同体积分数EA的洗脱剂进行洗脱,6种雌激素的回收率随洗脱剂极性的增加先增大后减小,当ACN与EA的体积比为4∶1时,回收率最高,达到89%,而当ACN与EA的体积比为5∶1时,6种雌激素的回收率下降,表明EA在洗脱过程中对回收率的影响较大。Fan等[14]认为这主要是由于EA的脂溶性可增加洗脱溶剂的渗透性,从而提高了提取效率。故采用MSPD前处理时,选用ACN-EA(4∶1, v/v)作为洗脱剂。
考察采用SPE萃取空白液态牛奶时不同洗脱剂对6种雌激素回收率的影响,结果见图2b。采用极性较大的ACN和MeOH时回收率只有70%;采用极性较低的DCM洗脱时回收率仅为50%;中等极性的EA可作为最佳洗脱剂,6种雌激素的平均回收率为76%;采用极性较EA稍大的ACN-EA(4∶1, v/v)洗脱时,6种雌激素的平均回收率为69%。故采用SPE前处理时,选用EA作为洗脱剂。
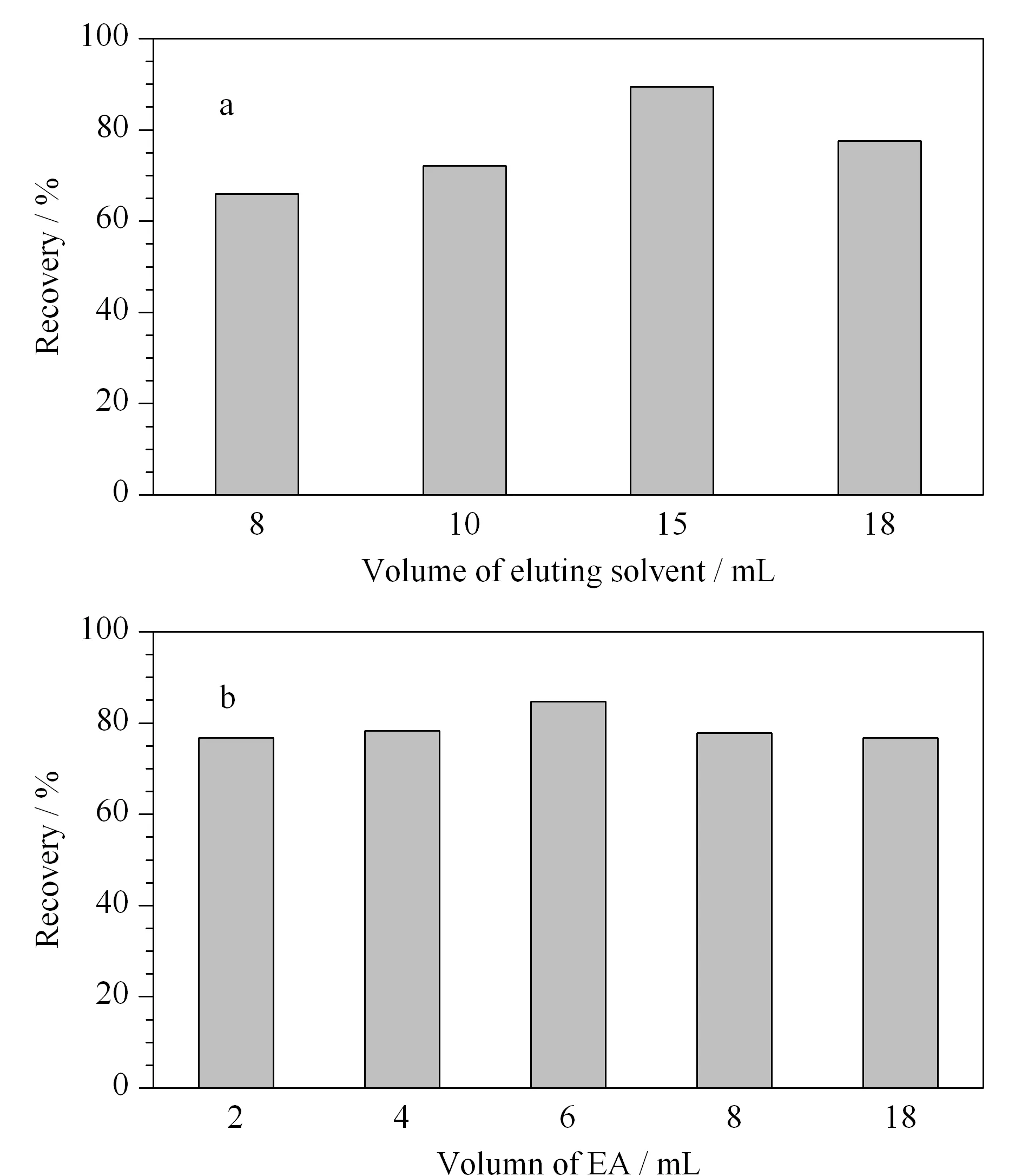
图 3 采用(a)MSPD和(b)SPE法时不同洗脱剂体积对6种雌激素回收率的影响Fig. 3 Effect of different volumes of the eluent on the recoveries of the six estrogens pretreated by (a) MSPD and (b) SPE
2.2.4MSPD与SPE洗脱剂体积的优化
对于MSPD过程中洗脱剂体积的优化,在硅胶吸附剂与样品质量比为1∶1的条件下采用8、10、15和18 mL的ACN-EA(4∶1, v/v)混合溶液进行洗脱,考察洗脱剂体积对6种雌激素回收率的影响(见图3a)。当洗脱剂的体积为8~15 mL时,6种雌激素的平均回收率逐渐增大,15 mL时达到89%;当洗脱剂体积增加至18 mL时,6种雌激素的平均回收率明显减小(77%),这可能是由于洗脱时间过长,对于基质中干扰组分也有一定的洗脱作用,致使目标化合物的回收率降低。故在MSPD处理过程中,洗脱剂ACN-EA (4∶1, v/v)的最佳用量为15 mL。
对于SPE过程中洗脱剂体积的优化,实验分别选择2、4、6、8和18 mL的EA作为洗脱剂。如图3b所示,当洗脱剂的体积为2~6 mL时,6种雌激素的回收率逐渐增加,继续增加洗脱剂体积,回收率逐渐降低。故在SPE处理过程中,洗脱剂EA的用量取6 mL为宜。
2.3 HPLC-Q-TRAP-MS条件的优化
实验采用胆固醇柱对6种雌激素进行分离分析,胆固醇柱对于结构类似的化合物,尤其是同分异构体或立体异构体具有良好的分离选择性。由图4的总离子流色谱图可见,互为立体异构体的17α-雌二醇和17-β雌二醇在胆固醇柱上的分离效果良好,其他雌激素类物质均达到基线分离。由于雌激素类物质大多含有酚羟基(除孕酮外),在ESI-模式下,产生的母离子为[M-H]-;孕酮和内标左炔诺孕酮在ESI+模式下形成[M+H]+母离子。在相应的离子化模式下,将6种雌激素标准溶液(1 mg/L)分别进行质谱参数的优化,优化结果见表2。
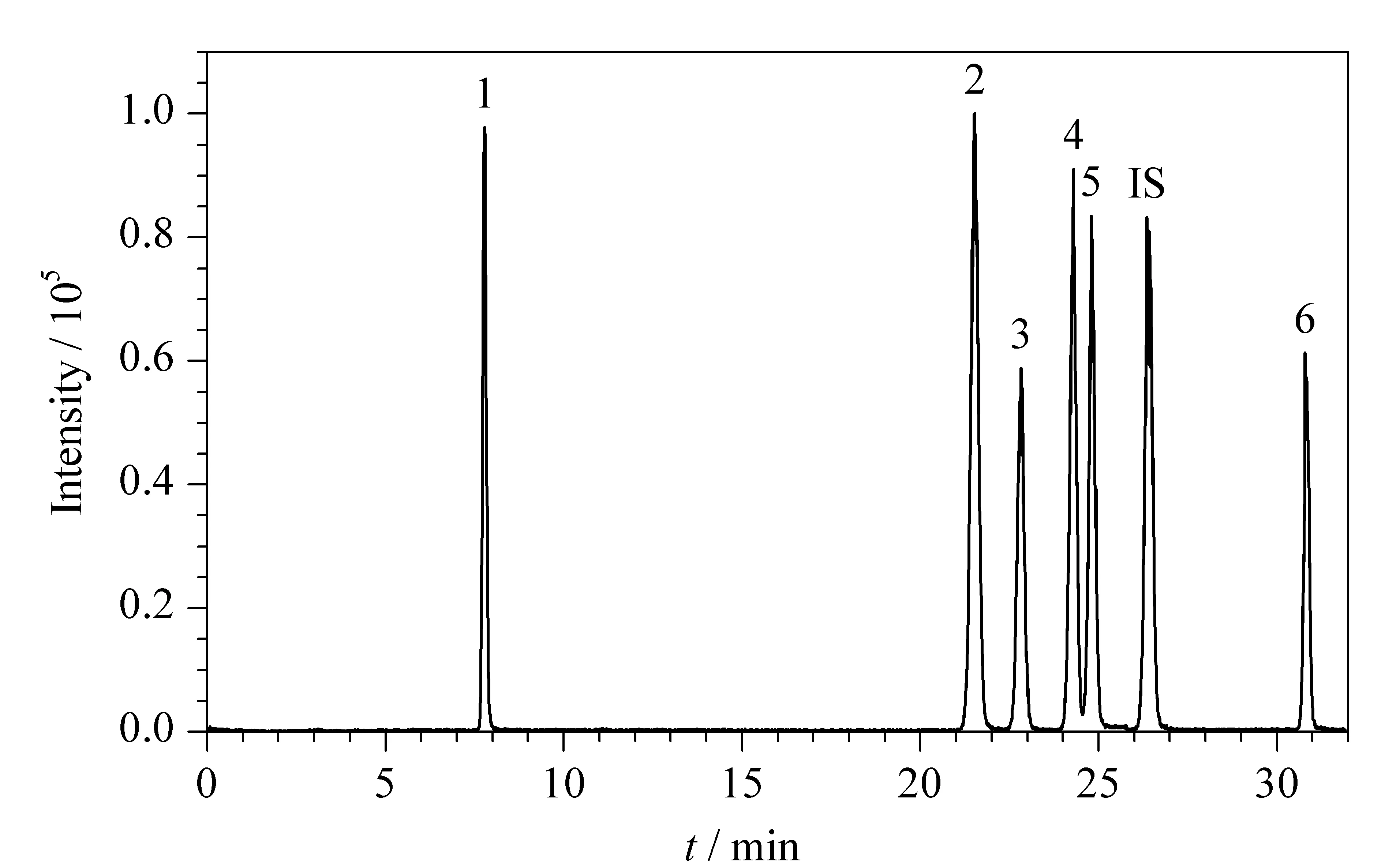
图 4 MRM模式下6种雌激素及内标(1 mg/L)的总离子流色谱图Fig. 4 Total ion chromatograms of the six estrogens and internal standard (1 mg/L) in MRM mode 1. E3; 2. 17α-E2; 3. 17β-E2; 4. E1; 5. EE; 6. P4; IS (internal standard): LNG.
2.4 方法学验证
2.4.1线性关系、基质效应和检出限
配制0.1~200 mg/L系列对照品混合标准溶液,内标的质量浓度均为10 mg/L。以各雌激素母离子与内标母离子色谱峰面积之比为纵坐标(y),各雌激素的质量浓度为横坐标(x,mg/L)绘制标准曲线(见表4)。在空白奶制品基质中添加相应浓度水平的雌激素,用内标法分别测定标准工作溶液及空白奶制品基质的标准曲线,以两者曲线斜率之比衡量基质效应。结果显示,MSPD处理下的6种雌激素的基质效应为89.4%~105.7%,而SPE处理下的6种雌激素的基质效应为86.7%~107.47%,表明两种预处理方法受样品基质中的杂质干扰均较小。以3倍和10倍信噪比(S/N)对应的样品中雌激素的质量浓度为方法的检出限(LOD)和定量限(LOQ)。由表4可见,6种雌激素在各自的线性范围内线性关系良好,相关系数(R2)均大于0.99;方法的LOD和LOQ分别为0.01~0.05 mg/L和0.05~0.10 mg/L。

表 4 6种雌激素的线性方程、线性范围、相关系数、检出限、定量限和基质效应Table 4 Linear equations, linear ranges, correlation coefficients (R2), limits of detection (LODs), limits of quantification (LOQs) and matrix effects of the six estrogens
y: parent ion peak area ratio of the estrogen to the internal standard;x: mass concentration, mg/L.
2.4.2回收率
在空白奶粉和液态牛奶中添加不同水平(1.0、5.0和10 mg/kg)的雌激素,分别按照MSPD和SPE的预处理方法对样品进行处理,每个水平重复3次,用内标法测定回收率,结果见表5。经MSPD法处理的6种雌激素的平均回收率为71.8%~106.0%(RSD为1.6%~9.2%,n=3),经SPE法处理的6种雌激素的平均回收率为70.3%~108.4%(RSD为2.0%~11.0%,n=3),说明方法的准确度高,重复性好。采用MSPD法处理时,EE的回收率为71.8%~77.3%,可能是因为硅胶作为中等极性吸附剂对疏水性较强的EE(Kow最大,见表1)的吸附作用较弱;用SPE萃取时,P4的回收率为70.3%~77.9%,或许是因为P4不含羟基,与SPE小柱的固定相之间无法通过有效的氢键作用增强其在柱上的保留,致使回收率偏低。

表 5 不同基质中6种雌激素的回收率和相对标准偏差(n=3)Table 5 Recoveries and relative standard deviations of the six estrogens in different matrices (n=3) %
2.4.3精密度与稳定性
选取低、中、高3个水平(0.15、2.0和160 mg/kg)的标准溶液,连续3 d分别对每个水平进样5次,测定仪器的日内精密度和日间精密度。结果表明,日内和日间精密度均不大于14.2%(见表6)。
对添加了不同水平(0.15和160 mg/kg)雌激素的空白奶粉和液态牛奶进行MSPD和SPE预处理,于0、2、4、8、12 h进样测定,考察0~12 h内雌激素的稳定性。结果显示,两种预处理方法处理样品后各雌激素含量的RSD均小于10%,表明在0~12 h内MSPD和SPE两种预处理方法下的样品均具有良好的稳定性。
2.5 实际样品的测定
应用所建立的方法,分别对市售的5种不同品牌的进口奶粉及3种进口牛奶样品中雌激素类药物残留量进行检测,均未检出上述6种雌激素。

表 6 6种雌激素在低、中、高添加水平下的日内和日间精密度(n=5)Table 6 Intra- and inter-day RSDs of the six estrogens at low, medium and high spiking levels (n=5) %
3 结论
本文分别用MSPD和SPE法纯化富集了日常奶制品中的6种雌激素,并针对不同基质选择了合适的预处理方法,同时优化了前处理条件。采用HPLC-Q-TRAP-MS系统进行分析,实现了快速、高效、高通量检测奶制品中雌激素残留。应用于实际样品的检测,也取得了令人满意的效果,或可为相关法律法规的制定及实际检测提供参考。
参考文献:
[1]Krieger N. Int J Epidemiol, 2008, 37: 627
[2]Penalver A, Pocurull E, BorrullF, et al. J Chromatogr A, 2002, 964: 153
[3]Xu X, Veenstra T D, Fox S D, et al. Anal Chem, 2005, 77: 6646
[4]Xu X, Roman J M, Issaq H J, et al. Anal Chem, 2007, 79: 7813
[5]Russo J, Russo I H. J Steroid Biochem Mol Biol, 2006, 102(1/5): 89
[6]Qin L Q, Wang P Y, Kaneko T, et al. Med Hypotheses, 2004, 62: 133
[7]Patisaul H B. P Nutr Soc, 2017, 76(2): 130
[8]Zaheer K, Akhtar M H. Critrev Food Sci, 2017, 57(6): 1280
[9]Yan W, Li Y, Zhao L X, et al. J Chromatogr A, 2009, 1216 (30): 7539
[10]EC 86/649
[11]GB 7916-2007
[12]Pérez-Fernández V, Morante-Zarcero S, Pérez-Quintanilla D. Electrophoresis, 2014, 35: 1666
[13]Li G L, Dong L H, Wang A H, et al. Food Sci Technol, 2014, 55: 355
[14]Fan Y B, Yin Y M, Jiang W B, et al. Food Chem, 2014, 142: 170
[15]Pan S D, He Q, Chen X H, et al. Chinese Journal of Chromatography, 2017, 35(9): 980
潘胜东, 何仟, 陈晓红, 等. 色谱, 2017, 35(9): 980
[16]Daeseleire E, Vandeputte R, Peteghem C V. Analyst, 1998, 123: 2595
[17]Fan H M, Li L Y. Chinese Journal of Ethnomedicine and Ethnopharmacy, 2011, 23: 69
范红梅, 李凌云. 中国民族民间医药, 2011, 23: 69
[18]Zakrzewski R, Ciesielski W. J Chromatogr B, 2003, 784: 283
[19]Lan H Z, Gan N, Pan D D, et al. J Chromatogr A, 2014, 1331: 10
[20]Giovanni D, Javier H, Herrera H, et al. Anal Bioanal Chem, 2016, 408: 7447
[21]Hua X G, Dai G M, Huang J J, et al. J Chromatogr A, 2010, 1217: 5875
[22]Luo H T, Huang X L, Wu H Q, et al. Chinese Journal of Chromatography, 2017, 35(8): 816
罗辉泰, 黄晓兰, 吴惠勤, 等. 色谱, 2017, 35(8): 816
[23]Barker S A. J Chromatogr A, 2000, 885: 115
[24]Zhang J, Wan H H, Zhang H. Chinese Journal of Chromatography, 2017, 35(9): 963
张婧, 万慧慧, 张华. 色谱, 2017, 35(9): 963
[25]Leon-Gonzalez M E, Rosales-Conrado N. J Chromatogr A, 2017, 1514: 88
[26]Fotouhi M, Seidi S, Shanehsaz M, et al. J Chromatogr A, 2017, 1504: 17
[27]Aufartováa J, Mahugo-Santanab C, Sosa-Ferrerab Z, et al. Anal Chim Acta, 2011, 704: 33
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02 08:22:54
中国特种设备安全(2021年12期)2021-04-26 14:37:00
时代英语·高一(2019年5期)2019-09-03 02:09:34
中成药(2018年6期)2018-07-11 03:01:32
中成药(2017年4期)2017-05-17 06:09:46
电测与仪表(2016年11期)2016-04-11 12:20:42
中国粮油学报(2016年5期)2016-01-23 02:45:06
核科学与工程(2015年3期)2015-09-26 11:58:24
电源技术(2015年5期)2015-08-22 11:18:28
天然产物研究与开发(2014年6期)2014-04-27 14:15:54
