
液相硝基甲烷分子振动特性的相干反斯托克斯拉曼散射光谱∗
2018-03-18 16:41彭亚晶孙爽宋云飞杨延强
物理学报 2018年2期
彭亚晶 孙爽 宋云飞 杨延强
1)(渤海大学物理系,锦州 121013)
2)(中国工程物理研究院流体物理研究所,绵阳 621900)
1 引 言
含能材料是一类含有爆炸性基团或含有可燃物和氧化剂的化合物或混合物.在一定外界能量的激发下,含能材料能够独立地进行化学反应并释放出能量[1−3].含能材料广泛应用于军事、航空航天以及工业等领域,在军事上可用于炮弹、导弹等,也可用于军事爆破等.含能材料的氧化剂和可燃物共聚一身,这使其具有敏感性和不稳定性.在外界热、力等能量刺激下,容易发生意外爆炸[4−9].因此,需要详细了解含能材料的起爆机理.
含能材料的分子反应发生在皮秒时间尺度和纳米空间区域.用飞秒激光研究含能材料反应的超快过程和过渡态近年来受到广泛关注.时间分辨相干反斯托克斯拉曼散射(CARS)光谱技术具有时间分辨率高、信号强度大、相干性好等优点,可以实现对原子、分子等物质微观结构的瞬态过程和瞬态效应的探测,广泛应用于化学、生物等分子反应动力学领域[10,11].因此,利用时间分辨CARS光谱技术探测含能材料分子内部结构的振动特性及其随时间的演化过程,可在原子分子水平洞悉分子内振动能量转移特性,对理解含能材料的微观反应机理具有重要作用[12−15].
目前,国内外科研机构已相继建立了CARS光谱技术,研究含能材料的分子振动能级结构及动态过程[15,16].美国华盛顿州立大学Winey等[17,18]利用时间分辨拉曼光谱技术研究了液体炸药硝基甲烷在冲击波作用下的反应动力学过程,获取了冲击作用下的分子结构变化、化学反应激发等重要信息.Namboodiri等[10]利用CARS光谱技术对液态硝基甲烷、硝基苯及二者的混合液进行了探测,分析了包含硝基基团的分子的振动动力学过程,表明飞秒时间分辨的CARS光谱和波数分辨的探测是分析电子基态动力学特性的一个有力工具.Cataliotti等[19]利用CARS光谱技术研究了液体硝基甲烷的振动弛豫特性,分析了温度和溶剂对振动失相和相关时间的影响,但研究的光谱范围较窄,只考虑了C—N伸缩振动的光谱.Hill等[20]利用CARS光谱研究了硝基甲烷的氢/氘同位素异构体光谱特性,得到了C—N伸缩振动的光谱形状,表明一个新的热带结构峰出现在C—N振动光谱附近.然而,Shkurinov等[21]随后的偏振CARS光谱研究表明,C—N伸缩振动附近的新峰结构并不是由热跃迁导致的,而是由硝基甲烷复合物所致.Dogariu和Pidwerbetsky[22]利用CARS光谱技术对环三亚甲基三硝胺(RDX)等含能材料的爆炸过程进行了实时跟踪探测,并分析了爆炸产物.美国Dlott研究组发展了新型高时空分辨的CARS光谱技术,研究纳米金属复合含能材料在激光驱动下的化学反应动力学、微区内反应的传播以及爆炸行为特性[23−25].他们的实验能够探测分子最基本的动力学过程,可实现与现有原子模拟方法的比较.尽管时间分辨激光光谱技术的应用促进了人们对含能材料微观反应特性的认识,但含能材料微观起爆机理尚无统一的定论,仍需在微观层面对其做进一步的研究.
为了能够在分子水平观察含能材料分子的动力学行为,本文搭建了以时间分辨CARS光谱为代表的非线性光谱系统,并对硝基甲烷这一典型的含能材料的分子振动特性进行了研究.获得了硝基甲烷分子几种主要振动模式的振动失相时间,给出了硝基甲烷分子化学键振动与热声子的耦合强弱特性,为理解热效应下硝基甲烷分子的初始化学反应机理提供了微观信息.
2 CARS光谱实验系统
图1所示为搭建的时间分辨CARS光谱实验系统.该系统采用的光源为钛蓝宝石主动锁模再生放大飞秒激光系统(Spectra Physics,美国),输出波长800 nm、脉宽130 fs、重复频率1000 Hz的脉冲激光.由光源输出的激光被分为三束,其中两束分别作为抽运光和探测光,对应的波矢为k1和k3;另外一束经衰减后聚焦在宝石晶体中产生超连续白光作为斯托克斯光,其波矢为k2.三束光经过各自的光学延迟线调整光程后,以空间折叠箱型结构聚焦在样品中间.在CARS过程中,抽运光和斯托克斯光共同作用于样品分子,激发分子的相干振动,分子的相干振动模会使探测光发生非弹性散射,从而产生CARS信号.扫描探测光的时间延迟即可探测分子相干振动弛豫的动力学过程.探测光的扫描时间间隔为25 fs,CARS光谱的时间分辨率约为130 fs,能够有效分辨7.7 THz(约260 cm−1)以下的振动态.CARS信号的产生是一个多光子过程,受动量守恒和能量守恒的限制.因此,信号出现在k1−k2+k3的相位匹配方向上.通过另一个透镜将信号准直后,经光纤收集进入光谱仪分光并由CCD探测.实验中采用的样品为分析纯液相硝基甲烷.硝基甲烷通常被称为液体炸药,是分子结构最简单的含硝基化合物,在含能材料的理论和实验研究中往往作为模型分子.

图1 CARS光谱实验系统Fig.1.CARS spectroscopic experimental system.
3 实验结果与分析
实验系统中采用宽带的超连续白光作为斯托克斯光,分子的所有振动模式原则上都能够同时被相干激发.但是,白光在介质中传播时存在色散效应,这导致白光中的不同频率成分不能严格地同时到达样品.因此,实验中需要适当调整白光的延迟时间以保证不同的振动模式能够被有效激发.在CARS光谱中,零时刻通常会出现一个最强的信号成分,该信号覆盖很宽的光谱范围.但该信号不与分子的任何振动模式相对应,而是非共振的电子背底信号,来源于电子云对外加光电场的瞬时响应.由于电子质量很小,因此非共振背底信号的弛豫时间非常短,与激光脉冲的脉宽在同一量级上(约百飞秒).非共振背底信号消失后出现的信号即为与不同分子振动模式相对应的CARS信号.
为了通过实验有效获得不同振动模式的CARS光谱,首先利用密度泛函的B3LYP/6-311++g**方法和基组获得硝基甲烷分子的拉曼光谱,如图2所示.对光谱中几个主要特征拉曼峰频率进行了校正[26]和指认,结果如表1所示.
由表1可见,频率在640 cm−1左右的拉曼峰主要由N—O键的对称弯曲振动所贡献;频率在897 cm−1左右的峰由C—N键的伸缩振动产生;频率在1355 cm−1附近的峰主要为C—H键的弯曲振动;频率在1384 cm−1附近的峰主要由N—O键的对称伸缩振动所产生;频率在2979 cm−1附近的拉曼峰则为C—H键的伸缩振动所致.该计算结果与文献[20,27]实验得到的655,917,1377,1400,2968 cm−1频率处振动情况基本一致.

表1 硝基甲烷分子的主要特征拉曼峰的频率归属Table 1.Frequency assignment of major Raman peaks of nitromethane molecule.
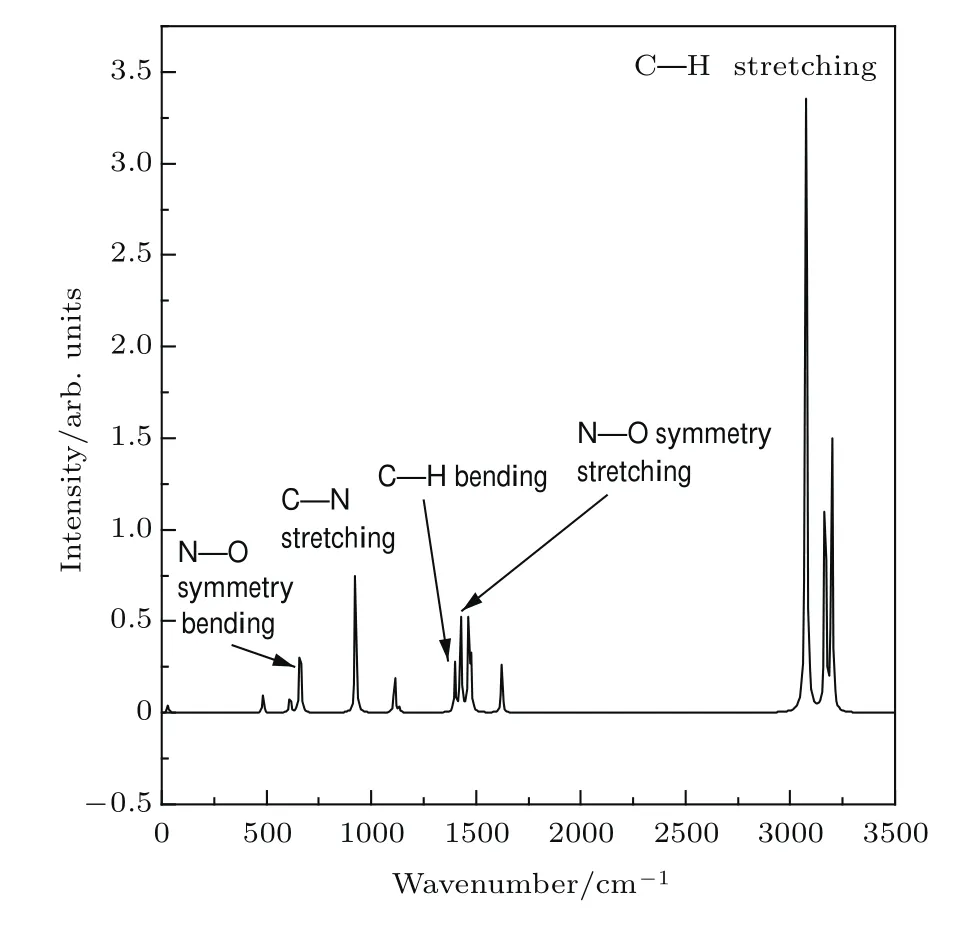
图2 计算获得的硝基甲烷分子的拉曼光谱Fig.2. Calculated Raman spectrum of the nitromethane molecule.
利用该实验系统研究液相硝基甲烷的相干振动弛豫动力学特性.首先将斯托克斯光的延迟时间调整到能够有效激发C—H键伸缩振动的位置,该振动模式位于3000 cm−1附近,得到的CARS光谱如图3(a)所示.但是,光谱中最明显的振动模式出现在917 cm−1处,对应C—N键的伸缩振动.尽管斯托克斯光的位置并不能使C—N键伸缩振动得到最有效的激发,但该模式仍然具有最高的强度.这是由于C—N键伸缩振动的拉曼活性远强于其他振动模式.图3(b)所示为3000 cm−1处C—H键的伸缩动力学曲线,该弛豫曲线反映了硝基甲烷的C—H键被相干激发后的失相过程.通过对该曲线的拟合可以得到C—H键伸缩振动的失相时间约为0.18 ps.
逐渐改变斯托克斯光的时间延迟,可以看到能量较低的振动模式在CARS光谱中依次出现.图4(a)表明随着时间延迟的改变,3000 cm−1处的C—H键伸缩振动信号逐渐消失,而位于1377和1404 cm−1处的振动信号强度明显增大,分别为硝基上N—O键的对称伸缩振动和甲基上C—H键的对称弯曲振动.图4(a)所示为CARS光谱中1400 cm−1处的弛豫曲线.图中出现了周期约为1.2 ps,即约为27 cm−1的弛豫曲线.该周期频率值与1377和1404 cm−1两个振动模式的频率之差相符,即出现了明显的拍频.拍频的出现是由于被探测的两个或多个振动模式的频率之差小于探测光本身的光谱带宽,导致两个振动模式的信号在光谱上相互叠加并发生干涉.拍频的频率通常等于发生干涉的两个振动模式的频率之差.因此,拍频的出现意味着N—O键的对称伸缩振动(1377 cm−1)和C—H键的对称弯曲振动(1404 cm−1)两个振动模式之间发生了干涉.在CARS实验中,所有被激发的振动模式都是相干的,即通过相干的光脉冲制备了分子体系的相干振动波包.波包随时间的演化特性是分子体系中重要的动力学参量.拍频振幅的衰减意味着波包正在坍缩,即形成波包的两个振动模式发生了退相干.由于图4所示数据信噪比并不是很高,且N—O键的对称伸缩振动和C—H键的对称弯曲振动两个振动模式的位置很近,所以可通过这两个振动模式的拍频振荡曲线拟合获得其振动失相时间.但由于N—O键的对称伸缩振动和C—H键的对称弯曲振动两个振动模式形成的拍频的波包失相很快,从实验数据中仅能分辨出两个较为明显的振荡周期.经初步拟合得到两个振动模式的失相时间均约为1.5 ps.

图3 (a)液相硝基甲烷的CARS光谱;(b)C—H键伸缩振动的弛豫曲线(红色实线为拟合曲线,用以获得失相时间)Fig.3.(a)CARS spectrum of liquid nitromethane;(b)relaxation curve of C—H stretching vibration(the red solid line is the fi tting curve to obtain the dephasing time).

图4 (a)液相硝基甲烷的CARS光谱;(b)N—O键伸缩振动和C—H键弯曲振动形成的振动波包(红色实线为拟合曲线,用以获得失相时间)Fig.4.(a)CARS spectrum of liquid nitromethane;(b)wave packet caused by N—O stretching vibration and C—H bending vibration(the red solid line is the fi tting curve to obtain the dephasing time).
继续改变斯托克斯光的时间延迟,将依次看到振动能量更低的两个振动模式的信号,即位于917 cm1处的C—N键伸缩振动和位于655 cm−1处的NO2中N—O键的对称弯曲振动,如图5所示.图5(a)所示CARS光谱的等高线图中,两个振动模式中间位置725 cm−1处出现了高频的量子拍.图5(b)所示为该量子拍的弛豫曲线.可见,量子拍周期约为0.125 ps,相应的频率为266 cm−1.该频率与两个被相干激发的振动模式917和655 cm−1的频率差(262 cm−1)基本相同.这证明该量子拍正是由C—N键伸缩振动模式和N—O键对称弯曲振动模式之间的相干导致的.
与前述硝基上N—O键的对称伸缩振动(1377 cm−1)和甲基上C—H键的对称弯曲振动(1404 cm−1)之间的量子拍不同,图5(b)中发生相干的C—N键伸缩振动(917 cm−1)和N—O键对称弯曲振动(655 cm−1)两个振动模式的间距较大.因此,除了两个信号的中间部分能看到拍频外,每个信号各自的弛豫过程也非常明显.通过对917和655 cm−1两个位置的弛豫曲线的拟合,可得到两个振动模式的失相时间分别为6.2和2.0 ps.这两个模式的失相时间比其他振动模式的失相时间长得多.同时,由于量子拍频率较高,能够明显看到两个模式之间的若干个拍频周期.

图5 (a)液相硝基甲烷的CARS光谱;(b)N—O键对称弯曲振动(655 cm−1),C—N键伸缩振动(917 cm−1),以及二者相干导致的拍频振动(725 cm−1)弛豫曲线(红色实线为拟合曲线,用以获得失相时间)Fig.5.(a)CARS spectrum of liquid nitromethane;(b)relaxation curves of N—O bending vibration(655 cm−1),C—N stretching vibration(917 cm−1),and their beat frequency vibration(725 cm−1)(the red solid line is the fi tting curve to obtain the dephasing time).
在常温的凝聚态体系中,导致失相的主要原因为随机的热碰撞,即热声子的散射使相干振动失相.尽管硝基甲烷分子间可能形成氢键,但文献[28]的研究表明,硝基甲烷分子间的氢键很弱.因此,分子间氢键对硝基甲烷振动失相的影响可以忽略.从这一角度来看,硝基甲烷分子的振动失相时间长意味着对应的振动与热声子的耦合作用较弱.比较硝基甲烷分子几种主要振动模式的振动失相时间可知,3000 cm−1处的C—H键伸缩振动失相时间只有0.18 ps,远小于代表甲基与硝基间伸缩振动的917 cm−1失相时间6.2 ps.然而热声子的能量通常在200 cm−1以下,因此,若单纯从能量的角度来看,频率越高的振动模式应该越不容易同热声子发生耦合,失相时间应该越长.而我们的实验结果表明,高频(3000 cm−1)C—H键伸缩振动失相时间最短,即该模式振动与热声子的耦合作用远强于低频917 cm−1C—N伸缩振动与热声子的耦合作用.可见,振动与热声子之间的耦合不仅受能量因素的影响,分子自身的结构以及分子间的相互作用方式都可能是影响耦合的因素.例如,在液态硝基甲烷中,由分子极性导致的分子间有序排列可能是C—N伸缩振动失相时间较长的主要原因.因此,对于硝基甲烷含能材料,在外界热加载情况下,C—H键的伸缩振动将有望最先被激发并引发初始化学反应.
4 结 论
利用CARS光谱实验系统,对液相硝基甲烷分子的相干振动动力学特性进行了研究.测得硝基甲烷分子几种主要振动模式的振动弛豫曲线,并获得了相应的振动失相时间.结果表明,低频917 cm−1处的C—N键伸缩振动的失相时间为6.2 ps,明显长于其他振动模式.而高频3000 cm−1处的C—H键伸缩振动的失相时间最短,为0.18 ps.这种低能量的C—N伸缩振动失相时间较长表明分子振动模式与热耦合强弱并不仅仅与振动能量有关,还会受到其他一些可能因素的影响,如分子结构和分子极性等.因此,C—H键伸缩振动对热声子的影响比C—N键伸缩振动对热声子的影响更敏感.在外界热加载的情况下,硝基甲烷的C—H键伸缩振动将有望最先被激发并引发初始化学反应.
[1]Peng Y J,Ye Y Q 2015Chemistry78 693(in Chinese)[彭亚晶,叶玉清 2015化学通报 78 693]
[2]Conner R W,Dlott D D 2012J.Phys.Chem.C116 14737
[3]Rossi C,Zhang K L,Estève D,Alphonse P 2007J.Microelectromech.Syst.16 919
[4]Peng Y J,Song Y F,Cai K D 2015Nanoaluminum Composite Energetic Materials(Beijing:Chemical Industry Press)p46(in Chinese)[彭亚晶,宋云飞,蔡克迪 2015纳米铝复合含能材料(北京:化学工业出版社)第46页]
[5]Liu Y,Jiang Y T,Zhang T L,Feng C G,Yang L 2015J.Therm.Anal.Calorim.119 659
[6]Pagoria P F,Lee G S,Mitchell A R,Schmidt R D 2002Thermochim.Acta384 187
[7]Sikder A,Sikder N 2004J.Hazard.Mater.112 1
[8]Badgujar D,Talawar M,Asthana S,Mahulikar P 2008J.Hazard.Mater.151 289
[9]Talawar M,Sivabalan R,Mukundan T,Muthurajan H,Sikder A,Gandhe B 2009J.Hazard.Mater.161 589
[10]Namboodiri V V,Ahmed M,Podagatlapalli G K,Singh A K 2015Proc.Indian Natl.Sci.Acad.81 525
[11]Wu H L,Song Y F,Yu G Y,Chen X L,Yang Y Q 2016J.Raman Spectrosc.47 1213
[12]Duan X H,Li W P,Pei C H,Zhou X Q 2013J.Mol.Model.19 3893
[13]Shan T,Thompson A P 2014J.Phys.Conf.Ser.500 172009
[14]Chu G B,Shui M,Song Y F,Xu T,Gu Y Q,Yang Y Q 2015J.Chem.Phys.28 49
[15]Cianetti S,Negrerie M,Vos M H,Martin J L,Kruglik S G 2004J.Am.Chem.Soc.126 13932
[16]Chan P Y,Kwok W M,Lam S K,Phillips D L 2005J.Am.Chem.Soc.127 8246
[17]Winey J M,Gupta Y M 1997J.Phys.Chem.B101 10733
[18]Winey J M,Duvall G E,Knudson M D,Gupta Y M 2000J.Chem.Phys.113 7492
[19]Cataliotti R S,Foggi P,Giorgini M G,Mariani L,Morresi A,Paliani G 1993J.Chem.Phys.98 4372
[20]Hill J R,Moore D S,Schmidt S C,Storm C B 1991J.Chem.Phys.95 3039
[21]Shkurinov A,Jonusauskast G,Rulliere C 1994J.Raman Spectrosc.25 359
[22]Dogariu A,Pidwerbetsky A 2012Lasers,Sources,and Related Photonic Devices,OSA Technical DigestpLM1B.2
[23]Guray T,Franken J,Hambir S A,Hare D E,Dlott D D 1997Phys.Rev.Letts.78 4585
[24]Yang Y,Hambir A A,Dlott D D 2002Shock Waves12 129
[25]Yang Y Q,Sun Z Y,Wang S F,Dlott D D 2003J.Phys.Chem.B107 4485
[26]Merrick J P,Moran D,Radom L 2007J.Phys.Chem.A111 11683
[27]Pangilinan G I,Gupta Y M 1994J.Phys.Chem.98 4522
[28]Megyes T,Bálint S,Grósz T,Radnai T,Bakó I 2007J.Chem.Phys.126 164507
猜你喜欢
汽车实用技术(2022年5期)2022-04-02
今日农业(2021年7期)2021-07-28
化工管理(2021年7期)2021-05-13
舰船科学技术(2021年12期)2021-03-29
军民两用技术与产品(2021年10期)2021-03-16
水上消防(2020年1期)2020-07-24
压电与声光(2019年5期)2019-10-22
疯狂英语·新读写(2018年3期)2018-11-29
中国农资(2016年1期)2016-12-01
火炸药学报(2014年5期)2014-03-20
