
基于MnO2纳米线-还原石墨烯复合修饰电极的多巴胺电化学检测
2018-03-14 00:57:38贺全国李广利邓培红刘晓鹏
分析化学 2018年3期
贺全国 梁 静 李广利* 邓培红 刘 军 刘晓鹏
1(湖南工业大学生命科学与化学学院, 株洲 412007) 2(衡阳师范学院化学与材料科学学院, 衡阳 421008)
1 引 言
多巴胺(DA)是一种重要的儿茶酚类神经递质,在中枢神经系统、肾、激素和心血管功能调节中起着重要作用[1]。目前,检测DA的方法包括色谱法[2,3]、荧光法[4,5]、电化学发光法[1,6]和电化学法[7,8]等,其中电化学法具有操作方便、灵敏度高、成本低、可原位检测等优势,已成为DA检测的常用方法。但DA、抗坏血酸(AA)、尿酸(UA)常共存于血液等生物样品中,且在裸电极上三者的氧化峰电位接近,容易对DA的检测造成干扰[9,10]。纳米复合材料修饰电极(金属纳米粒子修饰电极[6,11,12]、碳纳米管及其复合物修饰电极[13,14]、石墨烯及其复合物修饰电极[15~17]等)能提高电极的抗干扰性能和选择性,是解决这一问题的常用方法。
二氧化锰(MnO2)是一种用途广泛的过渡金属氧化物,具有低成本、低毒性、高纯度和优异的电化学性能等特点,广泛应用于可充电锂电池[18,19]、电催化氧还原[20,21]、电化学电容器[22,23]和化学或生物传感器[24~26]。由于具有高反应活性表面和高的比表面积,纳米MnO2材料在分析检测应用中具有突出优势[27]。一维纳米MnO2材料(纳米线)具有各向异性和高比表面积,是最有应用前景的电极修饰材料之一[28]。石墨烯自2004年发现以来,因其具有优异的导电性、高比表面积和良好的生物相容性等优势,广泛应用于电分析化学检测领域。MnO2/石墨烯复合材料由于兼具MnO2和石墨烯的优点,近年来成功用于电分析领域。Wang等[29]构建了MnO2纳米棒/石墨烯修饰电极,成功用于尿酸(UA)检测,且避免了抗坏血酸(AA)的干扰。Rani等[30]利用MnO2纳米片/多壁碳纳米管修饰电极成功检测H2O2。Zhang等[31]制备了不同形貌MnO2纳米粒子修饰碳糊电极,用于测定对乙酰氨基酚,响应峰电流较裸电极增加了约3倍。Wang等[32]利用超声法制备了MnO2/多壁碳纳米管复合纳米材料用于测定肼,线性范围为1.0×103~5.0×107mol, 响应时间小于2 s,检测限为0.2 mol。 但是,利用MnO2纳米线/石墨烯复合电极测定DA尚未见报道。
电化学还原方法是一种绿色方法,无需使用强还原剂,反应温和可控。本研究组曾利用电化学还原法构建了石墨烯及其复合物修饰的乙炔黑碳糊电极,成功用于色氨酸等生物活性物质和多酚A等添加剂的电化学检测[33~36]。本研究采用电化学还原方法制备MnO2纳米线-还原石墨烯复合修饰玻碳电极(MnO2-RGO/GCE)。系统优化了电化学还原条件和DA测定条件,研究DA在复合修饰电极的电化学行为。基于MnO2良好的电催化活性和还原石墨烯的高比面积、较强的表面吸附能力等优势,以及MnO2纳米线与还原石墨烯的协同作用,修饰电极对DA表现出了良好的检测性能。
2 实验部分
2.1 仪器与试剂
Hitachi S-3000N扫描电子显微镜(日本日立公司);JP-303E极谱分析仪(成都仪器厂);CHI 660E电化学工作站(上海辰华仪器厂)。
石墨粉、K3[Fe(CN)6]、(NH4)2S2O8、KMnO4、MnSO4·H2O等均为国产分析纯试剂。多巴胺(DA,Fluka 公司)。所有溶液均用超纯水(电阻率18.2 MΩ cm)配制。
2.2 实验方法
2.2.1氧化石墨烯的制备氧化石墨烯(GO)用改进的文献[37]方法制备,具体如下:将23 mL浓H2SO4冷却至0℃,加入0.5 g石墨粉和0.5 g NaNO3,机械搅拌均匀。冰水浴控制温度低于5℃,于搅拌下缓慢加入3 g KMnO4。升温至35℃,搅拌2 h,形成糊状体。温度控制在50℃以下,缓慢加入40 mL去离子水,升温至95℃反应0.5 h。加入100 mL去离子水,将以上混合溶液分几次搅拌下加入到20 mL 30% H2O2中,得到金黄色溶液。趁热抽滤,先用150 mL稀HCl(10%,V/V)清洗,再用150 mL去离子水清洗,将滤纸及其上面的固体在50℃下真空干燥,得到氧化石墨。 将上述100 mg 干燥的氧化石墨分散在100 mL 蒸馏水中,超声剥离2 h,离心两次,除去沉淀物,取上层清液,得浓度为0.95 mg/mL金黄色GO溶液。
2.2.2MnO2纳米线的制备称取0.008 mol MnSO4·H2O和0.015 mol (NH4)2S2O8,溶解于35 mL水中,120℃水热反应10 h。8000 r/min离心30 min,用水、乙醇清洗后,60℃下真空干燥,即得到MnO2纳米线。最后将其配成浓度为1 mg/mL的MnO2纳米线溶液备用。
2.2.3MnO2纳米线/石墨烯复合修饰电极的制备依次用1.0和0.3 μm的抛光粉打磨抛光玻碳电极(GCE)表面至镜面。将电极置于超纯水、无水乙醇、超纯水中各超声1 min,用高纯氮气吹干。然后将 1 mL MnO2纳米线溶液(1.0 mg/mL)分散于上述制备的20 mL GO溶液中,搅拌2 h,得MnO2-GO分散液。将5 μL MnO2-GO分散液滴涂到GCE表面,于红外灯下干燥,即得MnO2-GO/GCE修饰电极。最后在一定的还原电位和还原时间下,采用电化学还原GO,即制得修饰电极MnO2-RGO/GCE。
2.2.4电化学测定实验在磷酸盐缓冲液(PBS)中加入不同量的多巴胺,配制10 mL不同浓度DA溶液,测量前先通N2除氧。采用循环伏安法(CV)或二阶导数线性扫描伏安法进行电极表征或DA测定。电化学实验采用标准三电极体系:裸电极、RGO或MnO2-RGO修饰电极为工作电极,铂电极为辅助电极,饱和甘汞电极为参比电极。循环伏安法测定在CHI 660E 电化学工作站上进行,二阶导数线性扫描伏安法测定在JP-303E型极谱分析仪上进行。
3 结果与讨论
3.1 还原条件优化
电化学还原GO的电位一般为-1.5~-1.0 V。 DA(1×10-5mol/L)在不同还原电位制备的MnO2-RGO/GCE上的氧化峰电流如图1所示,当还原电位为-1.5 V时,氧化峰电流ipa最大。如图1B所示,还原时间60 ~300 s时,峰电流ipa随还原时间延长而增大,120 s达到最大值,随后还原时间延长,峰电流反而下降。选择制备修饰电极MnO2-RGO/GCE的最佳还原条件为还原电位-1.5 V,还原时间120 s。
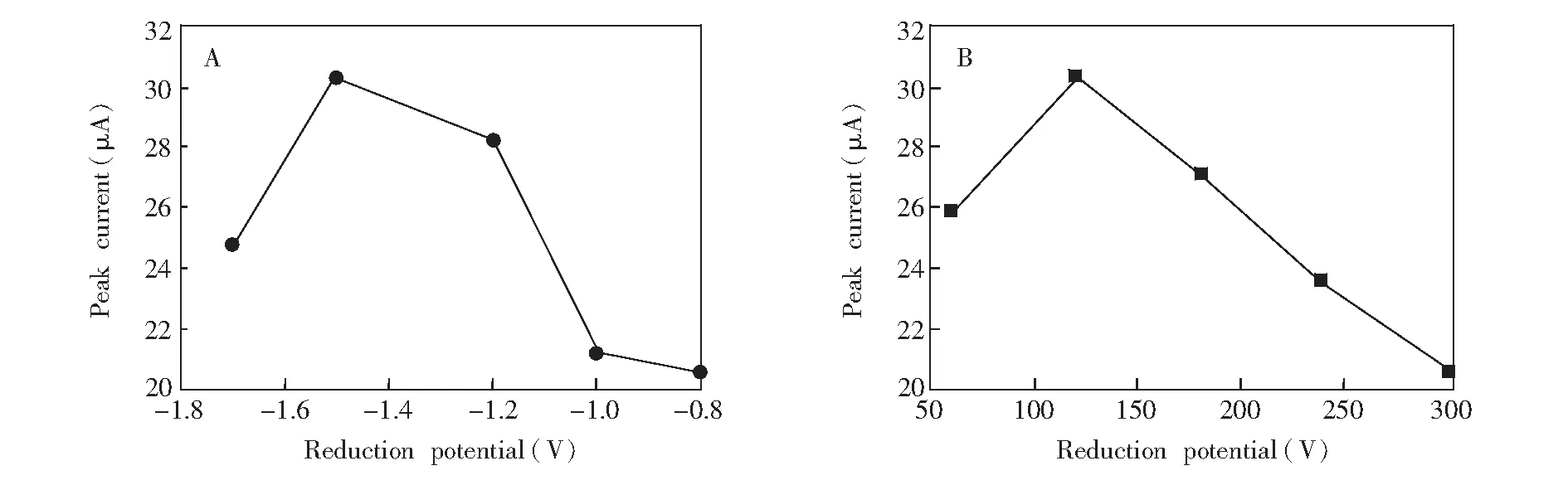
图1 电化学还原氧化石墨烯的还原电位(A)和还原时间(B)优化Fig.1 Optimization of reduction potential (A) and reduction time (B) for electrochemical reduction of graphene oxide (GO)

图2 RGO(A)、MnO2(B)及RGO-MnO2(C)SEM图谱Fig.2 Scanning electron microscopy (SEM) image of (A) reduced graphene oxide (RGO), (B) MnO2 nanowires, and (C) RGO-MnO2 nanocomposites
3.2 微观形貌表征
图2分别为RGO、MnO2纳米线和MnO2纳米线-RGO复合材料的SEM图谱。RGO为薄层弯曲绵延起伏状态(图2A)。图2B中MnO2纳米线呈分散均匀、尺寸一致的线状结构。图3为MnO2X射线粉末衍射图谱(XRD),在2θ=12.1°、18.0°、29.3°、37.5°、42.1°、50.1°、56.5°、60.5°和69.8°处出现明显的衍射峰,分别对应(110)、(200)、(310)、(211)、(301)、(411)、(600)、(521)和(541)位面(JSPDS44-0141,α-MnO2)。SEM和XRD的结果表明MnO2纳米线制备成功。从图2C可见,RGO的絮状薄层覆盖包裹MnO2纳米线,表明RGO与MnO2纳米线结合较好。这种RGO包覆MnO2纳米线结构,增大了复合材料的比表面积,有利于增加电化学活性面积和DA吸附。
3.3 DA在修饰电极上的电化学行为
不同电极在含1×10-5mol/L DA的0.1 mol/L PBS缓冲溶液中的电化学行为如图4所示。曲线a显示,DA在裸GCE上有宽的氧化还原峰宽,响应电峰流最小(ipa=2.434 μA,ipc=0.979 μA)。DA在RGO/GCE上的氧化还原峰电流有了较大提高(图4曲线b,ipa=22.10 μA,ipc=16.53 μA),可能是RGO大比表面积增加了电化学活性面积;RGO与待测物DA通过π-π键相互作用,增强了DA在电极表面的吸附能力。DA在MnO2-RGO/GCE的氧化还原峰峰形明显且尖锐(图4曲线c),氧化还原电流最大(ipa=34.55 μA,ipc=20.63 μA)。这主要是由于RGO与MnO2纳米线间的协同作用,MnO2具有电催化活性,可作为电子媒介体,促进电子在电极和DA之间快速传递;此外,RGO具有良好的导电性和高的比表面积,有效增强了电化学活性面积和DA吸附。
3.4 多巴胺测定条件优化
3.4.1底液pH值的影响图5为DA在MnO2-RGO/GCE上,在pH 2.0~5.5的PBS溶液中的循环伏安曲线图。随着pH值增加,氧化峰电位正移,还原峰电位负移。氧化峰电流与pH值的关系如图5B所示,在pH 2.0~3.0范围内,DA的氧化峰电流ipa随pH值增加而增大;当pH=3.0时,氧化峰电流ipa最大,随后pH值继续增加,峰电流ipa反而降低。故测定DA的pH选择3.0。
3.4.2扫速的影响图6A为不同扫速(30~300 mV/s)下,1×10-5mol/L DA(介质为pH=3.0 0.1 mol/L PBS)溶液在修饰电极MnO2-RGO/GCE上的循环伏安图。随着扫速增加,DA的氧化还原峰电流显著增大,但背景电流也随之增加(图6A)。由图6B可知,DA的氧化峰电流(ipa)与还原峰电流与扫速(v)在30~300 mV/s范围内呈良好的线性关系,线性方程分别为ipa=0.037v+0.0556(ipc)(R2=0.993),ipc=-0.036v+ 0.4393(R2=0.983)。这表明DA在修饰电极MnO2-RGO/GCE上的反应主要受吸附过程控制[4],因此后续实验采用富集方法增大电流响应信号。为了提高信噪比,减小背景电流,DA测定扫速选择100 mV/s。此外,随着扫速的增加,氧化峰电位正移,还原峰电位负移,说明反应为准可逆反应。峰电位与扫速的对数(lnv)呈良好的线性关系,线性方程分别为Epa=0.0328 lnv+0.484(R2=0.953),Epc=-0.0232 lnv+ 0.271(R2=0.952)。根据Lavrion方程[38]:
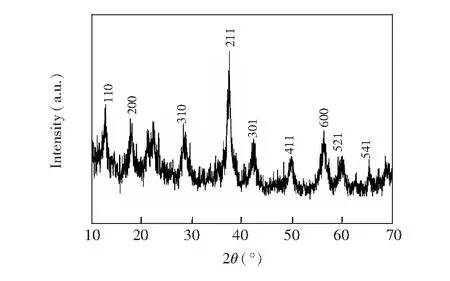
图3 MnO2纳米线XRD图谱Fig.3 X-ray diffraction (XRD) pattern of MnO2 nanowires
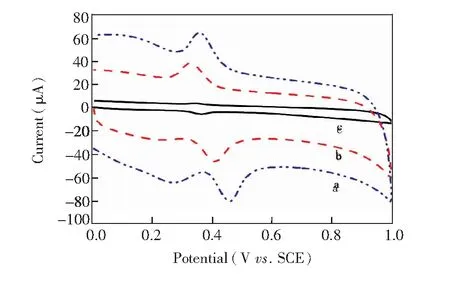
图4 1×10-5 mol/L DA在GCE(a),RGO/GCE(b),MnO2-RGO/GCE(c)上的循环伏安图Fig.4 Cyclicvoltammograms of 1×10-5 mol/L dopamine (DA) on different elctrodes in PBS buffer solution (0.1 mol/L pH 3.5). Scan rate: 100 mV/s. a, bare glassy carbon electrode (GCE); b, RGO/GCE; c, MnO2-RGO/GCE
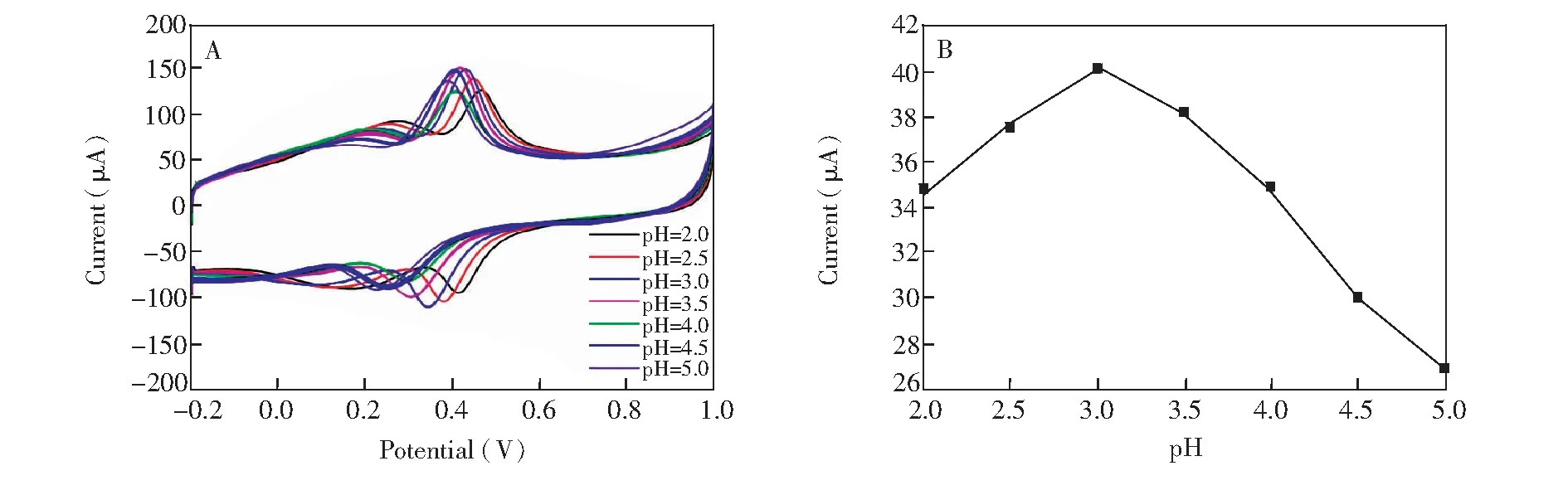
图5 (A)不同pH 值PBS溶液中 1×10-5 mol/L DA在MnO2-RGO/GCE上的循环伏安图;(B)氧化峰电流与pH 关系图Fig.5 (A) Cyclic voltammograms of 1×10-5 mol/L DA in PBS buffer solutions (0.1 mol/L) with different pH value; (B) Relationship between oxidation peak current and pH value
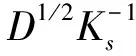
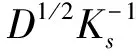
式中,Epa和Epc分别表示氧化峰电位和还原峰电位(V),v为扫描速率(V/s);α为电荷转移系数,ks为异相电子转移速率,D为扩散系数,n为电子转移数,T为开尔文温度,F为法拉第常数(96480 C/mol),R为摩尔气体常数(8.314 J/(mol·K))。结合公式(1)、(2)与上述Epa/Epc-lnv线性方程的斜率,即可得电荷转移系数α=0.6,电子转移数n=0.94≈1。DA氧化过程为等电子等质子过程,修饰电极MnO2-RGO/GCE测定DA属于单电子单质子快速传递过程,推测其机理为:首先,DA释放一电子成为半醌自由基;然后,半醌自由基歧化DA氧化物和DA。这与文献[38]报道的DA在GCE上的反应机理一致。推测的反应机理如下所示:
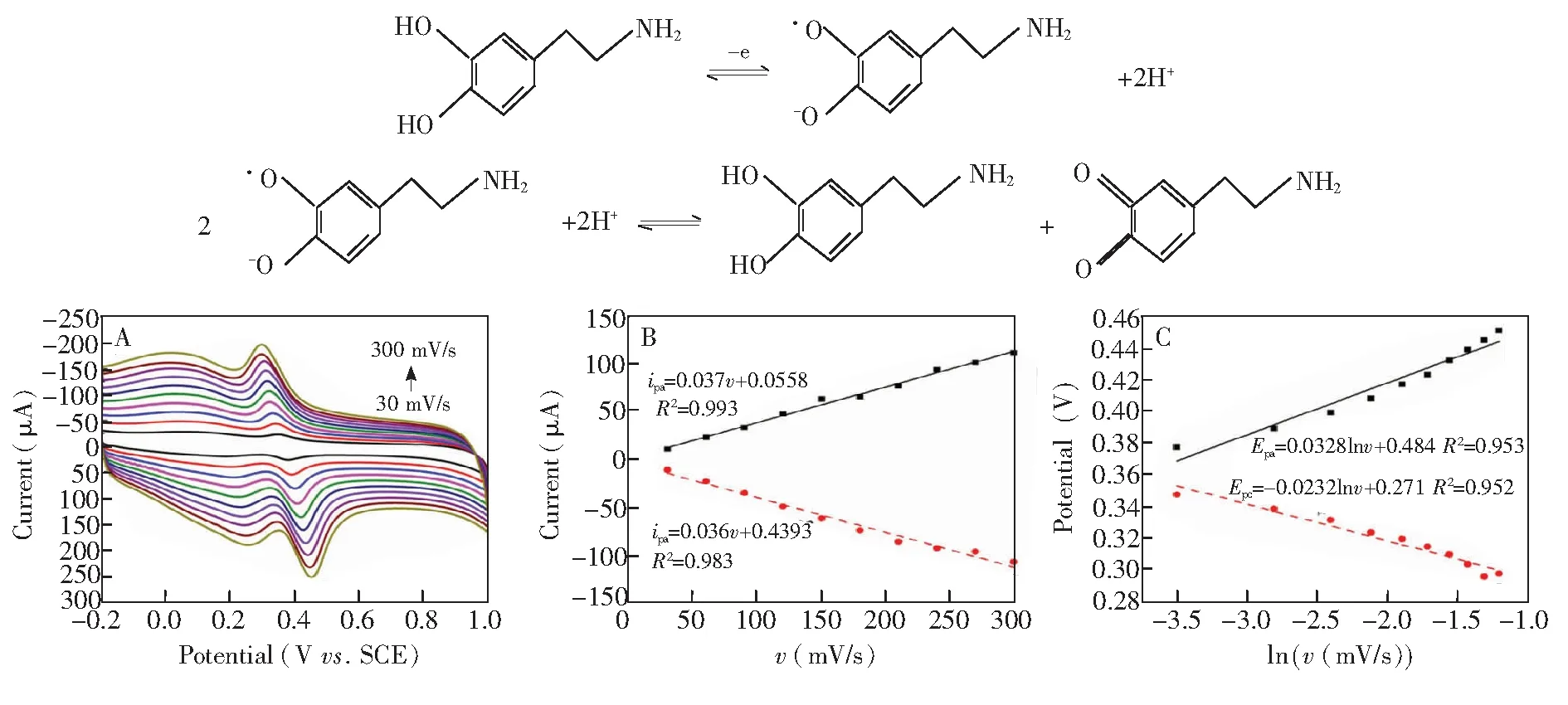
图6 (A)不同扫速下1×10-5 mol/L DA的循环伏安曲线;(B)DA氧化还原峰电流与扫速关系图;(C)DA氧化还原峰电位与扫速的对数关系图Fig.6 (A)Cyclic voltammograms of 1×10-5 mol/L DA with different scan rates; (B) Calibration curve between peak currents and scan rate; (C) Calibration curve between peak potentials and Napierian Logarithm of scan rate (lnv)
3.4.3富集条件优化不同富集电位(-0.4~0.2 V)下的溶出曲线(图7A)表明,峰电流大小与富集电位密切有关。在-0.4~-0.1 V范围内,随富集电位变正,峰电流增大;富集电位超过-0.1 V后,随电位升高,峰电流逐渐降低(图7A)。因此选择-0.1 V为测定DA的富集电位。图7B为峰电流与富集时间的关系图,随富集时间从30~150 s延长,峰电流增大;富集时间大于150 s后,峰电流基本保持不变,这说明DA吸附达到平衡。因此选择测定DA时的富集时间为150 s。
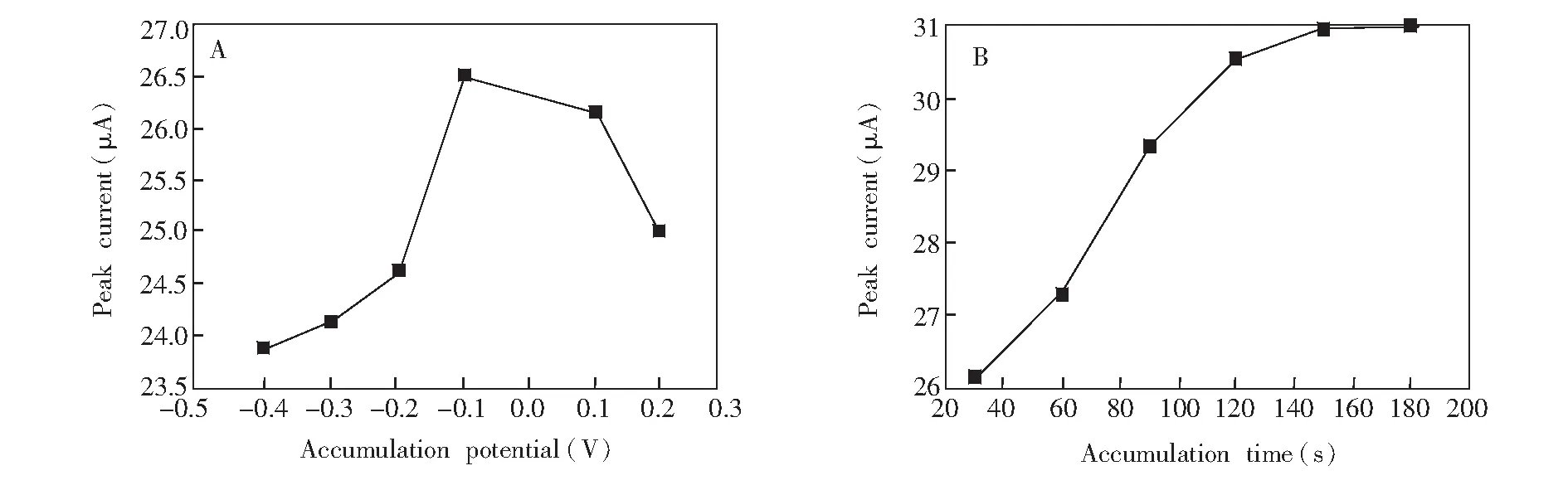
图7 富集电位(A)和富集时间(B)对1×10-5 mol/L DA氧化峰电流的影响Fig.7 Effect of accumulation potential (A) and accumulation time (B) on oxidation peak current of 1×10-5 mol/L DA
3.5 干扰实验
DA、抗坏血酸(AA)、尿酸(UA)共存于中枢系统细胞外液和血清中,在裸电极上三者的氧化峰电位接近,从而干扰DA的测定。线性溶出伏安法测定DA(2×10-5mol/L)与AA(1×10-5mol/L)、UA (1×10-5mol/L)共存时的结果如图8所示,DA、AA、UA三者的氧化峰电位分别为196、464和592 mV。氧化峰得到了明显分离,AA-DA氧化峰电位差ΔEp=268 mV,DA-UA之间的ΔEp=128 mV, 且与单独测定DA时的氧化峰电流未见明显减少。上述结果表明,石墨烯和MnO2纳米棒两者的协同效应提高了修饰电极的选择性和抗干扰能力。
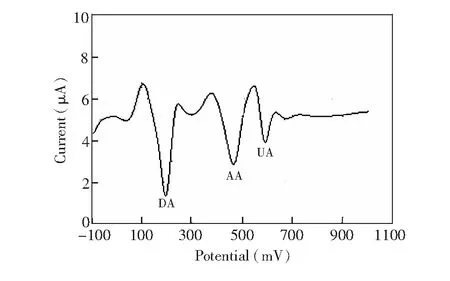
图8 DA(2×10-5 mol/L)、AA (1×10-5 mol/L)与 UA (1×10-5 mol/L) 混合样品在MnO2-RGO/GCE上的二阶导数线性扫描伏安图Fig.8 Second order derivative linear sweep voltammograms of mixture of DA (2×10-5 mol/L), AA (1×10-5 mol/L) and UA (1×10-5 mol/L) on MnO2-RGO/GCE
3.6 线性范围和检出限
由于二阶导数线性扫描伏安法相对循环伏安分辨率更好,测量精度更高,本方法采用二阶导数线性扫描伏安法测定多巴胺含量。在最佳测定条件下,采用线性伏安法对DA进行定量分析结果如图9A所示,当DA浓度为0.06~1.0 μmol/L时,氧化峰电流ipa均随浓度增大而增大,并呈良好的线性关系,线性方程为ipa=6.035c+ 8.581(R2=0.959);当DA浓度为1.0~80 μmol/L,浓度与峰电流同样呈线性关系,线性方程为 ipa= 0.421c+ 16.741(R2= 0.959)。检出限(S/N=3)为1.0 nmol/L。
重复5次测量2.0 μmol/L多巴胺的相对标准偏差(RSD)为3.4%,按相同方式制备4个MnO2-RGO/GCE,测定2.0 μmol/L DA响应峰电流的相对标准偏差为4.2%,表明电极制备和检测的重现性好。
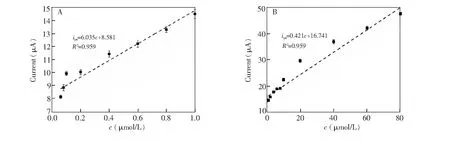
图9 DA浓度范围为0.06~1.0 μmol/L(A)和1.0~80.0 μmol/L(B),DA氧化峰电流ipa与浓度的关系曲线Fig.9 Relationship between oxide peak ipa and concentration of DA in the range of (A) 0.06-1.0 μmol/L and (B) 1.0-80.0 μmol/L
表1 本方法与文献报道检测DA方法的分析性能的比较 (n=4)
Table 1 Comparison of analytical performance between of method and other literature methods for detection of DA
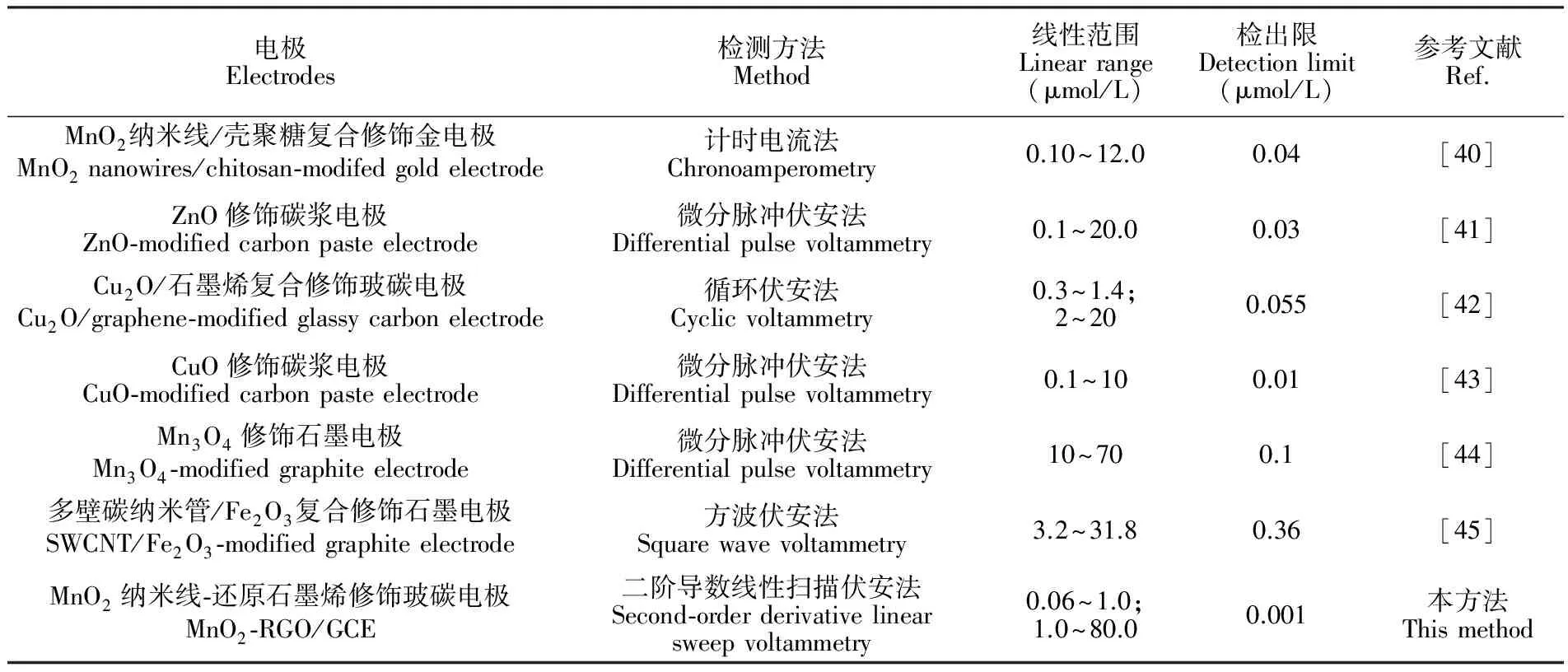
电极Electrodes检测方法Method线性范围Linearrange(μmol/L)检出限Detectionlimit(μmol/L)参考文献Ref.MnO2纳米线/壳聚糖复合修饰金电极MnO2nanowires/chitosan⁃modifedgoldelectrode计时电流法Chronoamperometry0.10~12.00.04[40]ZnO修饰碳浆电极ZnO⁃modifiedcarbonpasteelectrode微分脉冲伏安法Differentialpulsevoltammetry0.1~20.00.03[41]Cu2O/石墨烯复合修饰玻碳电极Cu2O/graphene⁃modifiedglassycarbonelectrode循环伏安法Cyclicvoltammetry0.3~1.4;2~200.055[42]CuO修饰碳浆电极CuO⁃modifiedcarbonpasteelectrode微分脉冲伏安法Differentialpulsevoltammetry0.1~100.01[43]Mn3O4修饰石墨电极Mn3O4⁃modifiedgraphiteelectrode微分脉冲伏安法Differentialpulsevoltammetry10~700.1[44]多壁碳纳米管/Fe2O3复合修饰石墨电极SWCNT/Fe2O3⁃modifiedgraphiteelectrode方波伏安法Squarewavevoltammetry3.2~31.80.36[45]MnO2纳米线⁃还原石墨烯修饰玻碳电极MnO2⁃RGO/GCE二阶导数线性扫描伏安法Second⁃orderderivativelinearsweepvoltammetry0.06~1.0;1.0~80.00.001本方法Thismethod
3.7 实际样品分析
将MnO2-RGO/GCE用于检测人血清中的DA。3份血清样品均未检出DA。采用标准加入法,测定50倍PBS稀释的人血清样品中的加标回收实验结果见表2,DA加标回收率为98.8%~102.0%,说明MnO2-RGO/GCE可用于血清中多巴胺的测定。
表2 人血清中多巴胺含量测定(n=4)
Table 2 Determination of DA in human blood serum samples (n=4)
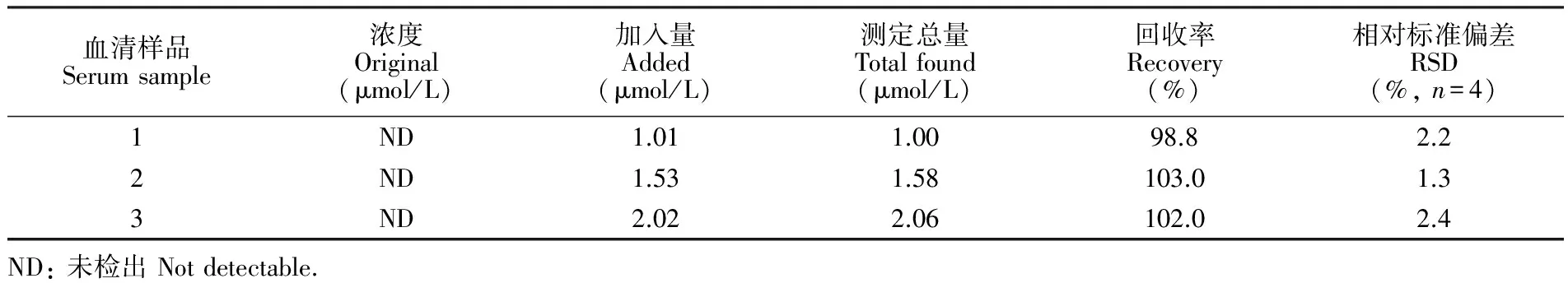
血清样品Serumsample浓度Original(μmol/L)加入量Added(μmol/L)测定总量Totalfound(μmol/L)回收率Recovery(%)相对标准偏差RSD(%,n=4)1ND1.011.0098.82.22ND1.531.58103.01.33ND2.022.06102.02.4ND:未检出Notdetectable.
1 Li L L, Liu H Y, Shen Y Y, Zhang J R, Zhu J J.Anal.Chem.,2011, 83(3): 661-665
2 Carrera V, Sabater E, Vilanova E, Sogorb M A.J.Chromatogr.B,2007, 847: 88-94
3 Guan C L, Ouyang J, Li Q L, Liu B H, Baeyens W R G.Talanta,2000, 50: 1197-1203
4 Wu H P, Cheng T L, Tseng W L.Langmuir,2007, 23: 7880-7885
5 Wang H Y, Sun Y, Tang B.Talanta,2002, 57: 899-907
6 Chen L F, Lu L L, Mo Y, Xu Z M, Xie S P, Yuan H Y, Xiao D, Choi M M F.Talanta,2011, 85: 56-62
7 Tian X Q, Cheng C M, Yuan H Y, Du J, Xiao D, Xie S P, Choi M M F.Talanta,2012, 93: 79-85
8 Zhang Y, Lin L L, Feng Z F, Zhou J Z, Lin Z H.Electrochim.Acta,2009, 55: 265-270
9 LI Chong, JIA Li-Ping, MA Rong-Na, JIA Wen-Li.Chem.J.ChineseUniversities,2015, 36(7): 1282-1290
李 冲, 贾丽萍, 马荣娜,贾文丽. 高等学校化学学报,2015, 36(7): 1282-1290
10 ZHANG Lei, LIN Xiang-Qin.Chem.J.ChineseUniversities,2003, 24(4): 591-594
张 雷, 林祥钦. 高等学校化学学报,2003, 24(4): 591-594
11 CHEN Dan, CAO Zhong, LIU Feng, WU Ling, XUN Yan, HE Jing-Lin, XIAO Zhong-Liang.ChineseJ.Anal.Chem.,2016, 44(10): 1593-1599
陈 丹, 曹 忠, 刘 峰, 吴 玲, 寻 艳, 何婧琳, 肖忠良. 分析化学,2016, 44(10): 1593-1599
12 Ulubay S, Dursun Z.Talanta,2010, 80(3): 1461-1466
13 Bi H Q, Li Y H, Liu S F, Guo P Z, Wei Z B, Lv C X, Zhang J Z, Zhao X S.Sens.ActuatorsB,2012, 171: 1132-1140
14 Yang S L, Li G, Yin Y L, Li JJ, Qu L B.J.Electroanal.Chem.,2013, 703: 45-51
15 Liu M L, Chen Q, Lai C L, Zhang Y Y, Deng J H, Li H T, Yao S Z.Biosens.Bioelectron.,2013, 48: 75-81
16 Lian Q W, He Z F, He Q, Luo A, Yan K W, Zhang D X, Lu X Q, Zhou X B.Anal.Chim.Acta,2014, 823: 32-39
17 Wu D, Li YY, Zhang Y, Wang P P, Wei Q, Du B.Electrochim.Acta,2014, 116: 244-249
18 Débart A, Paterson A J, Bao J L, Bruce P G.Angew.Chem.Int.Edit.,2008, 47: 4521-4524
19 Jiao F, Bruce P G.Adv.Mater.,2007, 19: 657-660
20 Cheng F Y, Su Y, Liang J, Tao Z L, Chen J.Chem.Mater.,2010, 22: 898-905
21 Gong K P, Yu P, Su L, Xiong S X, L.Q. Mao L Q.J.Phys.Chem.,2007, 111: 1882-1887
22 Liu R, Duay J, Lee S B.ACSNano,2010, 4: 4299-4307
23 Xu M W, Jia W, Bao S J, Su Z, Dong B.Electrochim.Acta,2010, 55: 5117-5122
24 Li Y L, Zhang J, Zhu H, Yang F, Yang X R.Electrochim.Acta,2010, 55: 5123-5128
25 Xu B, Ye M L, Yu Y X, Zhang W D.Anal.Chim.Acta,2010, 674: 20-26
26 Cao X, Wang N, Guo L.Sens.ActuatorsB,2009, 137: 710-714
27 Xu J J, Zhao W, Luo X L, Chen H Y.Chem.Commun.,2005, 6(6): 792-794
28 Bai Y H, Xu J J, Chen H Y.Biosens.Bioelectron.,2009, 24: 2985-2990
29 Wang Z H, Tang J, Zhang F F, Xia J F, Shi G Y, Xia Y Z, Xia L H, Qin L C.Int.J.Electrochem.Sci.,2013, 8(7): 9967-9976
30 Rani K K, Devasenathipathy R, Wang S F, Yang C.Ionics,2017, (7-8): 1-8
31 Zhang L C, Liu Z H,Lv H, Tang X H, Ooi K.J.Phys.Chem.C,2007, 111(24): 8418-8423
32 Wang M F, Wang C, Wang G F, Zhang W, Bin F.Electroanalysis,2010, 22(10): 1123-1129
33 Deng P H,Xu Z F, Zeng R Y, Ding C X,FoodChem.,2015, 180: 156-163
34 Deng P H,Xu Z F, Kuang Y F.J.Electroanal.Chem.,2013, 707: 7-14
35 Deng P H,Xu Z F, Feng Y L.Mater.Sci.Eng.C,2014, 35: 54-60
36 Deng P H,Xu Z F, Li J H.Microchim.Acta,2014, 181: 1077-1084
37 Hummers W S,Offeman R E.J.Am.Chem.Soc.,1958, 80: 1339-1340
38 Laviron E J.Electroanal.Chem.,1979, 101(1) : 19-28
39 YE Bao-Xian, LI Feng-Ju, ZHANG Jun, JIN Bao-Hui.Chem.Res.,2003, 14(1): 44-46
冶保献, 李风菊, 张 俊, 靳保辉. 化学研究,2003, 14(1): 44-46
40 Huang Y, Cheng C, Tian X, Zheng B, Li Y, Yuan H, Xiao D, Choi M M F.Electrochim.Acta,2013, 89: 832-839
41 Reddy S,Swamy B E K, Chandrashekar B N, Chitravathi S, Jayadevappa H.Anal.Bioanal.Electrochem.,2012, 4: 186-196
42 Zhang F, Li Y,Gu Y E, Wang Z, Wang C.Microchim.Acta,2011, 173(1-2): 103-109
43 Reddy S,Swamy B E K, Jayadevappa H.Electrochim.Acta,2012, 61: 78-86
44 Gao W, Ye S, Shao M.J.Phys.Chem.Solids,2011, 72(9): 1027-1031
45 Adekunle A S, Agboola B O, Pillay J, Ozoemena K I.Sens.ActuatorsB,2010, 148(1): 93-102
猜你喜欢
欣漾(2024年2期)2024-04-27 12:03:09
疯狂英语·新悦读(2023年2期)2023-10-12 14:42:47
中学生数理化·中考版(2022年12期)2022-02-16 07:36:52
原子与分子物理学报(2021年2期)2021-03-29 07:31:10
疯狂英语·新读写(2020年3期)2020-06-06 09:05:58
时代英语·高一(2019年5期)2019-09-03 02:09:34
材料科学与工程学报(2016年4期)2017-01-15 13:35:33
材料科学与工程学报(2016年2期)2017-01-15 13:34:35
电子制作(2016年23期)2016-05-17 03:53:41
肇庆学院学报(2016年5期)2016-03-11 18:09:20
