
Contemplation on some cyclic N8isomers-A DFT treatment
2018-03-12 08:03Lemirker
Defence Technology 2018年1期
Lemi Türker
Middle East Technical University,Department of Chemistry,Üniversiteler,Eskis¸ehir Yolu No:1,06800 Çankaya,Ankara,Turkey
1.Introduction
One of the most abundant elements in nature is nitrogen.It forms the highlystable N2molecule in its elemental state.However,in contrast to this form of it,polynitrogen compounds(PNC),comprising only nitrogen atoms are rare,and no molecular crystal made of these compounds has been prepared yet[1].
Various allotropic modifications of nitrogen,namely,compounds consisting only of nitrogen atoms(of the form N2,N3,N4,etc.),are classified as polynitrogen compounds.They are considered as promising candidates of clean(green)high energy density materials(HEDM)because they produce N2gas only and have high energy content[2-5].
In order to seek novel high energy density compounds(HEDCs)having no air pollution,attempts of scientists have been focused on the concept of polynitrogen compounds,which attract significant interest for propulsion or explosive applications.Through the years,potential candidates of polynitrogen compounds have been predicted by the theoreticians since the early 1990s and lots of systematic and great efforts have been undertaken in order to synthesize any of them[6-16].
It is believed that use of polynitrogen compounds will allow solid rocket propellants to compete in terms of energetic efficiency with liquid propellants[2,17].According to theoretical calculations and(still scarce)experimental data,the polynitrogen compounds are characterized by high enthalpy of formation(2-5 kcal/g)and sufficiently high density in the condensed phase(2-4 g/cm3)[17].It has been theoretically estimated that the use of polynitrogen compounds can provide a specific impulse of 350-500 s with material density in a range of 2.0-3.9 g/cm3[2].
In general polynitrogen molecules are expected to release large amounts of energy when they decompose into the very stable N2molecules.Due to that fact,these structures are potentially promising molecules as high-energy-density materials(HEDM)[18-20].In recent years,pure polynitrogen molecules have been particularly attractive,among the different energetic nitrogen compounds(such as nitrates,ammonia,nitramines,azides,polyazides and so on),not only because of both the expected high energy density but also N2is the sole product of their decomposition,which is inert,non-toxic and not a greenhouse gas[1].
So far,the quest for HEDMs based on nitrogen atoms has produced several theoretical articles.Only one solid-state material containing a N+5cation and a gas phase N5-anion were reported experimentally in addition to the well-known N-3anion and N3radical.Other species,such as N4,were only observed as short-lived transients.Several topical reviews have portrayed the difficulty in preparing all nitrogen compounds[21-24].Christe and coworkers recently reported two breakthroughs in the field:the synthesis of the N+5cation in a salt[7]and of the cyclo-N-5anion in the gas phase[25].According to theoretical calculations,the cation is V-shaped whereas the anion is cyclic[23].
Hirshberg et al.,made use of PW-DFT with the PBE-D[26,27]functional to investigate the relative thermodynamic stability,the enthalpies of the N8solid and cg-N form(cubic gauche)at pressures up to 50 GPa[1].Their work reveals the possibility of existence of such a molecular solids,consisting of N8molecules although it is metastable even at ambient pressure.Their calculations predict a conceptually interesting new material on condition that,if it could be prepared,as HEDM it may find some applications.
In the present study,various cyclic N8structures have been considered within the constraints of density functional theory.
2.Method of calculation
Geometry optimizations of all the structures leading to energy minima were initially achieved by using MM2 method followed by semi-empirical PM3 self-consistent fields molecular orbital(SCF MO)method[28,29]at the restricted level[30,31].Subsequent optimizations were achieved at Hartree-Fock level using various basis sets.Then,geometry optimizations were managed within the framework of density functional theory(DFT,B3LYP)[32,33]finally at the level of 6-311++G(d,p)(restricted closed-shell)[30].Additionally,UB3LYP/6-311++G(d,p)and B3LYP/CC-PVTZ level of calculations were performed for energies.The exchange term of B3LYP consists of hybrid Hartree-Fock and local spin density(LSD)exchange functions with Becke's gradient correlation to LSD exchange[33,34].Note that the correlation term of B3LYP consists of the Vosko,Wilk,Nusair(VWN3)local correlation functional[35]and Lee,Yang,Parr(LYP)correlation correction functional[36].The vibrational analyses were also done.The total electronic energies are corrected for the zero point vibrational energy(ZPE).The normal mode analysis for each structure yielded no imaginary frequencies for the 3N-6 vibrational degrees of freedom,whereNis the number of atoms in the system.This indicates that the structure of each molecule corresponds to at least a local minimum on the potential energy surface.All these calculations were done by using the Spartan 06 package program[37].The NICS(0)values were calculated(B3LYP/6-311++G(d,p))by the use of Gaussian 03 package program[38].
3.Results and discussion
Cyclic only-nitrogen structures are interesting not only because of their extra ring-strain energy as compared to their acyclic counterparts but also due to some other properties.Note that in the present treatment yet-non-existing structures considered are called isomers rather than allotropes.
Fig.1 shows the optimized structures of N8isomers(singlet states)obtained at the level of B3LYP/6-311++G(d,p)calculations.In the present treatise six(see Fig.1)N8isomers are considered.Bicyclic N8structure shown below was found to be unstable in the singlet and triplet states.Therefore,it has not been considered furthermore.
3.1.Some properties and energies
Table 1 shows some properties of the isomers.Except 1 and 3 all the others have zero dipole moment.Note that 2 is thetransform(of 4-membered rings)of 3 and it does not have any dipole moment but 1(has a 4-membered ring)and 3 have.So the 4-membered ring(s)contributes into the total asymmetry to develop dipole moment via individual bond dipoles.In thetransstructure-2,the direction of dipole moment indicates that two of the 4-membered rings are non-identical in terms of electron population.
The structures included in Table 1 all have either low or zero dipole moments.Fig.1 also displays the direction of the dipole moments.
Tables 2-4 show the total electronic energies,zero point vibrational energies(ZPE)and the corrected total electronic energies.The B3LYP/6-311++G(d,p)and B3LYP/cc-PVTZ level of calculations yield the stability order of 4>6>5>1>2>3 where the values of 4 and 6 are the same.The UB3LYP/6-311++G(d,p)level of calculations exhibit the stability order of 4>6>5>1=2>3.In the case of B3LYP/ccPVTZ level of calculations,the stability order mimics the order of B3LYP/6-311++G(d,p)level of calculations.Note that in every case the energies of 4 and 6 are very close to each other.The reason for it will be given in the sections below.
The stability orders indicate that it is adversely affected by the number(s)of 4 m-membered rings.Although,structure-4 is a 4 mmembered ring(m=2)it is the most stable one among the group.Note that it does not have any dipole moment.Structures-5 and 6 do not have any 4-memebered rings.The direction of dipole moment in 1 indicates that the 4-membered ring is somewhat deprived of electrons in the favor of the 6-membered ring.At first sight,a question arises whether some aromatic character having 6π-electrons(see the following NICS section)associates with the hexagonal ring system and the 4-membered ring having a localized(N=N)double bond which is distant from the fusion site of the rings exists.
Fig.2 shows the optimized triplet state of N8isomers considered.As seen in the figure some bonds are highly elongated indicating some fragmentation.Table 5 displays the various energies of the structures in the triplet state.Note that for the decomposed structures,the energies in the table stand for the composite(fragmented)systems.Although system 1 has the lowest energy,actually it is the decomposed one.Structure 1 and 5 seem to be splitted into N2fragments.Structures-2 and-3 have partially broken skeletons(see Fig.2).Whereas structures 4 and 6 keep their integrity having reasonable bond lengths.They show high mutual similarity in the singlet and triplet state geometry and energy.
The triplet state stability order is 1>5>6>4>2>3.Otherwise mentioned below the structures considered are all in their singlet states.
3.2.Heats of formation
The heats of formation(ΔHf0)values for the N8species(singlet state)considered are obtained by using T1 method[39,40].The T1 method is a little bit less accurate than the expensive G3(MP2)method.For the comparison purpose,T1 and G3(MP2)results obtained for structure 1 and presently they have been found to be 303.94 kcal/mol and 304.67 kcal/mol,respectively(-0.09%deviation).Table 6 shows the heats of formation values for the N8species(singlet state).The order of endothermicity is 3>2>1>5>4>6.So structures-4 and 6 are distinguished as electronically the most stable and least endothermic ones in the group.
Structure-3 which possesses thecisconfiguration of4-membered rings is the most endothermic one followed by 2 which hastransconfiguration.So the 4-membered rings,depending on their configuration in the structure contribute somewhat into the endothermicity.Since bond energies are the prime contributors of thermal nature of molecules in general,in structures 1-6 which are all composed of nitrogen,single or double bond character of nitrogen bonds and conjugation are to be blamed for the spectrum of heats of formation values in Table 6.
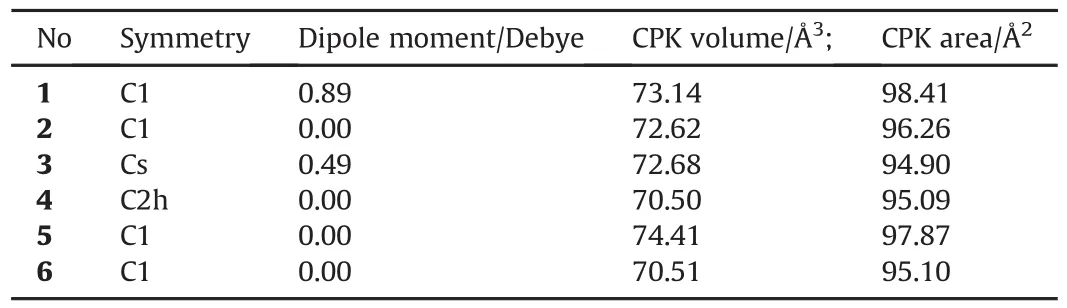
Table 1Some properties of the singlet structures considered(B3LYP/6-311++G(d,p)).

Table 2Various energies of the N8singlet state structures(B3LYP/6-311++G(d,p)).

Table 3Various energies of the N8singlet state structures(UB3LYP/6-311++G(d,p)).
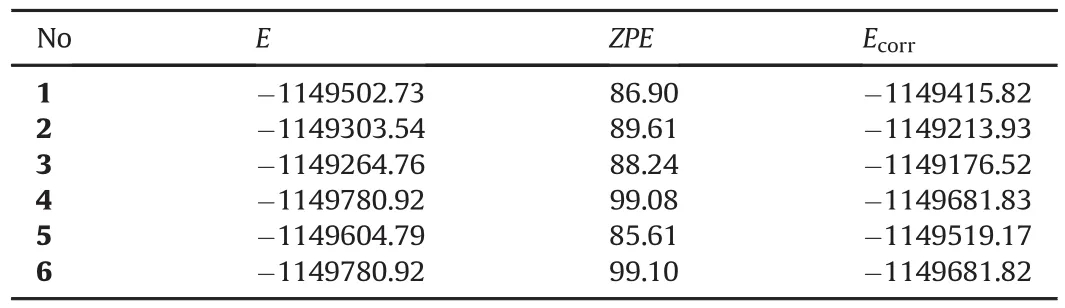
Table 4Various energies of the N8singlet state structures(B3LYP/cc-PVTZ).
3.3.Bond lengths
Figs.3 and 4 display the numbering of the atoms and bond lengths(B3LYP/6-311++G(d,p))in the structures,respectively.In structure-1 the hexagonal moiety(have 6π-electrons)shows bond alternation.Although,the direction of the dipole moment is from4-memebered ring to 6-memebered one,the last one does not exhibit any comparable bond lengths which is the characteristic feature of perfect aromatic systems like benzene(see also the NICS section below).Note that bond alternation is the characteristic feature of annulenic carbocyclic compounds.
For azapentalene(analogous to structure-6)Noyman et al.,reported CCSD/cc-PVDZ-calculated bond lenghts for structure having(C2V)symmetry as 1.324 Å (N2-N6),1.339 Å (N6-N7),1.324 Å (N7-N8)(see Fig.3 for numbering of the atoms)[41].The results are very close to the present values.
In structure-4,N1-N8 distance(apparently there is no bond there)is the same with the bond length there in structure-6(N1-N2 bond in Fig.4)which is 1.337 Å.Note that the experimentally determined bond lengths for N-N and N=N are 1.449 Å(hydrazine)and 1.219-1.254 Å(azo compounds),respectively[42].

Table 5Various energies of the N8triplet state structures.
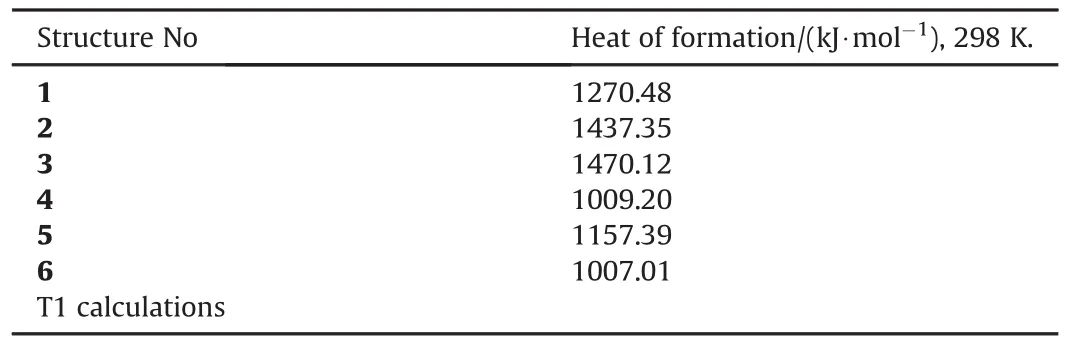
Table 6Heat of formation values of the structures.
3.4.Mulliken and L¨owdin bond orders for structures 4 and 6
Structures-4 and 6 exhibit a striking similarity.Table 7 shows the Mulliken and L¨owdin bond orders for structures-4 and 6.The numerical results indicate that these two structures have the same types of bonds.Moreover,there exists a bond between N1 and N8 atoms of structure-4 which has the same length in structure 6.Note that N1 and N8 atoms in structure-4 stand for N1 and N2 of structure-6,respectively(see Fig.3).
In Table 7 all the atom numbers of structure-6 have been adjusted based on the numbering of structure-4 for the purpose of easier and better comparison.
3.5.Electrostatic charges
Fig.5 shows the electrostatic charges(esu)on the atoms of N8isomers.Note that the largest charge accumulation in 1 occurs at the fusion points.A similar situation happens in structure-6.
In structure-4,although apparently there is no common bond between the pentagonal moieties to be considered as fused,the charge distribution is very similar to it is in 6(see also sections below).
3.6.NICS
Since nitrogens in the structures possess lone-pair electrons,their involvement in the cyclic conjugation should be checked out.For this purpose NICS(0)[43,44]values have been calculated at the B3LYP/6-311++G(d,p).
Table 8 shows the NICS(0)values for the rings in N8isomers.In the calculations,the rings are considered as having cyclic conjugation by the participation of nitrogen lone pairs in a suitable manner.Ring-A(6-membered)and Ring-B in structure-1 are planar but highly antiaromatic(especially Ring-B).In structures-2 and-3 the 4-memebered rings in the same structure exhibit different character.Even,Ring-A of structure-2 exhibits slight aromaticity(or nonaromaticity)having a negative NICS value.Note that Ring-A of 2 is somewhat depleted of electrons,thus the dipole moment tail originates from there(see Fig.1).This cationic nature should be the cause of negative NICS value of Ring-A,namely it is not a pure 4π-system(characterized withantiaromaticity)but less.These differences in the NICS values might arise from slight structural and electronic variations in the rings.
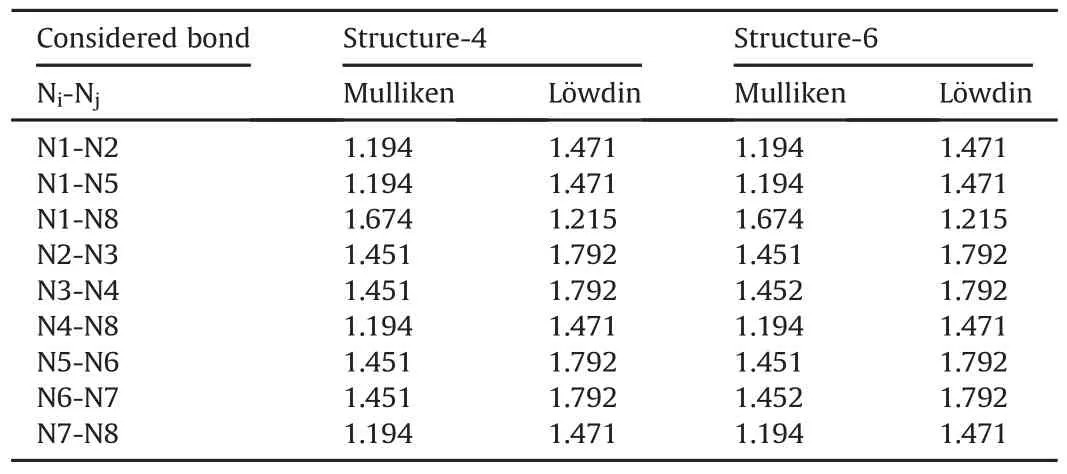
Table 7Mulliken and L¨owdin bond orders for structure-4 and 6.

Table 8NICS value of the rings(singlet structures).
Structure-4 is interesting.It is planar and apparently,it is a 8πsystem,suggesting antiaromatic nature but the NICS values for its 8π-system or its embedded 4π-system over the five nitrogens atoms(pseudo 5-membered ring)yield highly aromatic character.Thus probably a homoaromatic[45]occurrence takes place in spite of the fact that there is no bond at the fusion points of pseudo 5-membered rings complete the conjugation over.Similar to structure-4,structure-6 exhibits an aromatic character.

Table 9HOMO,LUMO energies and interfrontier energy gaps of the singlet structures considered.
Structure 5(tube form of N8)has not been included in Table 8,because it is not suitable for aromaticity treatment,thus for NICS calculations due to i)the whole structure is not planar;ii)any part of it cannot be considered for homoaromaticity because of the long distance between N1 and N4 or N5 and N8(see Fig.3 for the numbering of atoms)positions to construct a hypothetical planar rings.Note that these distances are 2.571 Å and 2.551 Å where as the distance between N1 and N8(see Fig.3)in structure 4(where homoaromaticity is considered)are 1.337 Å and 1.331 Å depending on the basis sets used in the present treatment.
3.7.Spectra
Fig.6 shows the calculated IR(upper axis)and UV(lower axis)spectra of structure 1-6.The IR spectra having 4-membered ring(s)has/have some peaks at about 1500 cm-1characteristic of N=N stretching of the 4-membered ring(s).Structure-5 also has peaks at 1500 cm-1(1526-1600 cm-1,various N=N stretchings).All theconsidered structures,with the exception of 5,possess a strong peak at 1000 cm-1(various N-N stretchings).Note that IR spectra of 4 and 6 have high resemblance.

Table 10Molecular orbital energies for structures 4 and 6.
The time-dependent density functional theory(TD-DFT)yields the UV-VIS spectra of the present structures shown in Fig.6.The figure indicates that structures 1 and 5 should absorb in some part of visible region as well,whereas 4 and 6 spectra have been con fined to UV region solely.The great resemblance existing between 4 and 6 is also observed in their spectra.
3.8.Molecular orbital energies
Table 9 shows the highest occupied,lowest unoccupied molecular orbital energies(εHOMO,εLUMO,respectively)and the frontier molecular energy gaps(namely εHOMO- εLUMO).The HOMO energy order is 4< 6<2< 3<1< 5 whereas the LUMO energy order is 4> 6>2> 3>5> 1.These energy orders dictate the FMO gap as 5< 1<3<2<6< 4.Structures 4 and 6 are characterized with the lowest lying HOMO and highest lying LUMO energies.Thus,their FMO gap is greater than the others in the group.
Note that electron attracting factors lower both the HOMO and LUMO energy levels whereas electron donating ones raise up both of them[46].such kind of situation seems to be mutually operative in 4 and 6 to yield the resultant energies of the frontier molecular orbitals(HOMO and LUMO)which implies some polar resonance structures possible where the charges located in such a symmetrical manner that no dipole moment of 4 and 6 exists(see section 3.9).
Table 10 displays the molecular orbital energies(up to four digits)of curiously resembled structures-4 and 6.The data reveal that their similarity so far indicated also present in their molecular orbital energies.Also note that their HOMO and LUMO patterns pair wise are the same(see Fig.7).
3.9.Possible conversion of N8from monocyclic to bicyclic structure
Fig.8 shows a possible route to conversion of 4 to 6 via 4a(middle structure in Fig.8).Although,4a is a charge separated structure it is more stable than 4.Moreover,4a and 6 are characterized with the same total electronic energy (B3LYP/6-311++G(d,p)).The energy values of 4,4a and 6 are-114355.06,-1149573.46 and-1149573.46 kJ/mol,respectively.The activation energy for the conversion of 4 to 4a is just 150.18 kJ/mol.Note that 6 is more charge separated resonance structure than 4a but they are degenerate in terms of the stability.The underlying reason is most probably the aromaticity of the 5-membered rings.
Fig.9 shows the bond lengths(Å)of structures 4 and 6(B3LYP/cc-PVTZ).As seen in Fig.4(which displays the bond lengths of the singlet structures considered at the level of B3LYP/6311++G(d,p))this level of calculations also indicate the very high resemblance between structures 4 and 6.In 4 the distance between atoms 1 and 8 is 1.332 Å.
Structure-4 has a 4 m-type π-skeleton and classically such a conjugated monocyclic planar system is antiaromatic by the Hückel considerations.Whereas structure-6 has two aromatic rings(6πsystem).So the conversion of 4 to 6 should be a favorable process.The aromatic stabilization energy liberated in the process possibly counterbalance the required energy fort he conversion.However,in the optimization process,the computer program most probably conceive 4(a monocyclic structure)as 6(a bicyclic structure).Therefore,there exists a great resemblance in between them.
3.10.Impulse values of N8structures considered
Since it is believed that use of polynitrogen compounds will allow solid rocket propellants to compete in terms of energetic efficiency with liquid propellants[2,17]and for propellants,the material's potential is best measured by its specific impulse,Isp,presently that property has been estimated for structures 1-6.The specific impulse in units of seconds can be approximated with the following equation[5].
Ispfor monocyclic N8is reported as 400 s(CCSD/cc-PVDZ level of calculation)[5,41].Table 11 tabulates the heats of formation and the specific impulse values.The order ofIspvalues is 3>2>1>5>4>6.The order indicates that presence of 4-membered rings highly raise theIspvalues whereas the aromaticity decreases.
Note that the present heats of formation values have been calculated by T1 recipe which closely reproduce heats of formation values calculated from G3(MP2).The later one has been developed for thermochemical calculations.The T1 recipe operates by replacing the large basis set MP2 calculation by a dual basis set RI-MP2 calculation and replace the QCISD(T)calculation and vibrational frequency calculation by an empirical correction based on atom and bond counts[37].

Table 11The heat of formation values and specific impulses of cyclic N8structures.
4.Conclusions
The considered polynitrogen structures of mono and bicyclic N8isomers have been found to be stable but highly endothermic.Structures 4 and 6 have been found to be the least endothermic and most stable ones.Isomer 4 and 6 showed great resemblance to each other in terms of many respects.The resemblance between them is independent of basis set keeping the same level of calculations in both cases.Although,the apparent structure of 4 should associate with an antiaromatic nature,NICS calculations reveals that both structures-4 and 6 are aromatic in character.The resemblance between these antiaromatic(supposedly)and aromatic pair has been attributed to existing homoaromaticity in 4 within the constrains of DFT.They are characterized with very comparable impulse values too.
[1]Hirshberg B,Gerber RB,Krylov AI.Calculations predict a stable molecular crystal of N8.Nat Chem 2014;6:52-6.
[2]Zarko VE.Searching for ways to create energetic materials based on polynitrogen compounds(review).Combust Explos Shock Waves 2010;46:121-31.
[3]Klap¨otke TM,Harcourt RD.The interconversion of N12to N8and two equivalents of N2.J Mol Struct(theochem)2001;541:237-42.
[4]Smirnov A,Lempert D,Pivina T,Khakimov D.Basic characteristics for estimation polynitrogen compounds efficiency.Central Eur J Energetic Mater 2011;8:233-47.
[5]Wilson KJ,Perera SA,Bartlett RJ,Watts JD.Stabilization of pseudo-benzene N6ring with oxygen.J Phys Chem A 2001;105:7693-9.
[6]Peng L,Lai W,Chang H,Li Y,Li H,Yang W,Wang Y,Wang B,Xue Y.Density functional theoretical study of polynitrogen compounds N5+Y-(Y=B(CF3)4,BF4,PF6and B(N3)4).Chin J Chem 2012;30:639-43.
[7]Christe KO,Wilson WW,Sheehy JA,Boatz JA.N5+:a novel homoleptic polynitrogen ion as a high energy density material.Angew Chem Int Ed 1999;38:2004-9.
[8]Vij A,Wilson WW,Vij V,Tham FS,Sheehy JA,Christe KO.Polynitrogen chemistry.Synthesis,characterization,and crystal structure of surprisingly stable fluoroantimonate salts of N5+.J Am Chem Soc 2001;123:6308-813.
[9]Wilson WW,Vij A,Vij V,Bernhardt E,Christe KO.Polynitrogen chemistry:preparation and characterization of(N5)2SnF6,N5SnF5,and N5B(CF3)4.Chem Eur J 2003;9:2840-4.
[10]Christe KO,Vij A.AFRL-PR-ED-TR-2004-0041.History of the AFRL/USC DARPA program on polynitrogen chemistry,vol.2;October,2004.
[11]Hiraoka K,Yamabe S.Stabilities of the N3+(N2)ncluster ions withn=1-11.Chem Phys Lett 1989;154:139-42.
[12]Pyykk¨o P,Runeber N.Ab initiostudies of bonding trends:Part 9.The dicyanamide-carbon suboxide-dicyanoether-cyanogen azide isoelectronic series.J Mol Struct(Theochem)1991;234:279-90.
[13]Xu WG,Li GL,Wang LJ,Li QS.Ab initioand density functional theory study of the mechanism of synthesis of the Nq5cation.Chem Phys Lett 1999;314:300-6.
[14]Nguyen MT,Ha TK.Theoretical study of the pentanitrogen cation N5+.Chem Phys Lett 2000;317:135-41.
[15]Wang X,Hu HR,Tian A,Wong NB,Chien SH,Li WK.An isomeric study of N5+,N5,and N-5:a Gaussian-3 investigation.Chem Phys Lett 2000;329:483-9.
[16]Netzloff HM,Cordon MS,Christ K,Wilson WW,Vij A,Boatz JA.On the existence of FN5,a theoretical and experimental study.J Phys Chem A 2003;107:6638-47.
[17]Talawar MB,Sivabalan R,Aasthana SN,Singh H.Novel ultrahigh energy materials.Combust Expl Shock Waves 2005;41:264-77.
[18]Hammert A,Klap¨otke TM,Schwerdtfeger P.Azoylpentazoles as high energy materials,a computational study.Chem Eur J 2003;9:5511-9.
[19]Christe K.Recent advances in the chemistry of N+5,N-5and high-oxygen compounds.Prop Explos Pyrotech 2007;32:194-204.
[20]Najafpour J,Nejad CF,Shafiee GM,Peykani MK.How does electron delocalization affect the electronic energy?A survey of neutral poly-nitrogen clusters.Comput Theor Chem 2011;974:86-91.
[21]Cacace F,de Petris G,Troiani A.Experimental detection of tetranitrogen.Science 2002;295:480-1.
[22]Nguyen MT.Polynitrogen compounds 1.Structure and stability of N4and N5systems.Coord Chem Rev 2003;244:93-113.
[23]Samartzis PC,Wodtke AM.All-nitrogen chemistry:how far are we from N60?Int Rev Phys Chem 2006;25:527-52.
[24]Klapotke TM.New nitrogen-rich high explosives,in:high energy density materials.Struct Bond 2007;125:85-121.
[25]Vij A,Pavlovich JG,Wilson WW,Vij V,Christe KO.Experimental detection of the pentaazacyclopentadienide(pentazolate)anion,cyclo-N5-.Angew Chem Int Ed 2002;41:3051-4.
[26]Perdew JP,Burke K,Ernzerhof M.Generalized gradient approximation made,simple.Phys Rev Lett 1996;77:3865-8.
[27]Grimme S.Accurate description of van der Waals complexes by density functional theory including empirical corrections.J Comput Chem 2004;25:1463-73.
[28]Stewart JJP.Optimization of parameters for semi empirical methods I.Method J Comput Chem 1989;10:209-20.
[29]Stewart JJP.Optimization of parameters for semi empirical methods II.Appl J Comput Chem 1989;10:221-64.
[30]Leach AR.Molecular modeling.Essex.Longman;1997.
[31]Fletcher P.Practical methods of optimization.New York:Wiley;1990.
[32]Kohn W,Sham L.Self-consistent equations including exchange and correlation effects.J Phys Rev 1965;140:1133-8.
[33]Parr RG,Yang W.Density functional theory of atoms and molecules.London:Oxford University Press;1989.
[34]Becke AD.Density-functional exchange-energy approximation with correct asymptotic behavior.Phys Rev A 1988;38:3098-100.
[35]Vosko SH,Vilk L,Nusair M.Accurate spin-dependent electron liquid correlation energies for local spin density calculations:a critical analysis.Can J Phys 1980;58:1200-11.
[36]Lee C,Yang W,Parr RG.Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density.Phys Rev B 1988;37:785-9.
[37]SPARTAN 06.Irvine CA,USA:Wave function Inc.;2006.
[38]Frisch MJ,Trucks GW,Schlegel HB,Scuseria GE,Robb MA,Cheeseman JR,Montgomery Jr JA,Vreven T,Kudin KN,Burant JC,Millam JM,Iyengar SS,Tomasi J,Barone V,Mennucci B,Cossi M,Scalmani G,Rega N,Petersson GA,Nakatsuji H,Hada M,Ehara M,Toyota K,Fukuda R,Hasegawa J,Ishida M,Nakajima T,Honda Y,Kitao O,Nakai H,Klene M,Li X,Knox JE,Hratchian HP,Cross JB,Bakken V,Adamo C,Jaramillo J,Gomperts R,Stratmann RE,Yazyev O,Austin AJ,Cami R,Pomelli C,Ochterski JW,Ayala PY,Morokuma K,Voth GA,Salvador P,Dannenberg JJ,Zakrzewski VG,Dapprich S,Daniels AD,Strain MC,Farkas O,Malick DK,Rabuck AD,Raghavachari K,Foresman JB,Ortiz JV,Cui Q,Baboul AG,Clifford S,Cioslowski J,Stefanov BB,Liu G,Liashenko A,Piskorz P,KomaromiI,Martin RL,FoxDJ,Keith T,Al-Laham MA,PengCY,Nanayakkara A,Challacombe M,Gill PMW,Johnson B,Chen W,Wong MW,Gonzalez C,Pople JA.Gaussian 03.Revision C.02.Wallingford CT:Gaussian,Inc.;2004.
[39]Ohlinger WS,Klunzinger PE,Deppmeier BJ,Hehre WJ.Efficient calculation of heats of formation.J Phys Chem A ACS Publ 2009;113:2165-75.
[40]Curtiss La,Raghavachari K,Redfern PC,Rassolov V,Pople JA.Gaussian-3(G3)Theory for molecules containing first and second-row atoms.J Chem Phys 1998;109:7764-76.
[41]Noyman M,Zilberg S,Haas Y.Stability of polynitrogen compounds:the importance of separating the σ and π electron systems.J Phys Chem A 2009;113:7376-82.
[42]Vilkov LV,Mastryukov VS,Sadova NI.Determination of the geometrical structure of free molecules.Moscow:Mir Pub;1983.
[43]Pulay P,Hinton JF,Wolinski K.Nuclear magnetic shieldings and molecular structure.In:Tossel JA,editor.NATO ASI series C,vol.386.Netherlands:Kluwer;1993.p.243.
[44]Hehre WJ,Radom L,Schleyer PR,Pople JA.Ab Initio molecular orbital theory.New York:Wiley;1986.
[45]Minkin VI,Glukhovtsav MN,Simkin BY.Aromaticity and antiaromaticity.New York:Wiley;1994.
[46]Fleming I.Frontier orbitals and organic chemical reactions.London:Wiley;1976.

- Defence Technology的其它文章
- Approximate ballistics formulas for spherical pellets in free flight
- An experimental study on shock wave mitigation capability of polyurea and shear thickening fluid based suspension pads
- Cold metal transfer(CMT)technology-An overview
- Numerical and experimental study of wave shaper effects on detonation wave front
- Microstructure,properties and hot workability of M300 grade maraging steel
- Modification of RDX and HMX crystals in procedure of solvent/anti-solvent by statistical methods of Taguchi analysis design and MLR technique