
Modulation of mitochondrial bioenergetics as a therapeutic strategy in Alzheimer’s disease
2018-03-06 03:46IsaacOnyango
中国神经再生研究(英文版) 2018年1期
Isaac G. Onyango*
Gencia Biotechnology, Charlottesville, VA, USA
Introduction
Alzheimer’s disease (AD) is a major health issue affecting over 46 million people worldwide. Without therapeutic breakthroughs, the number could reach 75 million in 2030 and exceed 131.5 million by 2050 (www.alz.co.uk/worldreport2016). The current annual global economic cost associated with managing dementia exceeds US$818 billion, and AD will become a trillion-dollar disease by 2018 (https://www.alz.co.uk/research/WorldAlzheimerReport2016.pdf).
AD is categorized into two major forms: sporadic AD (sAD)and familial AD (fAD) with < 10% of AD cases being familial(Thinakaran, 1999) and showing autosomal-dominant transmission within affected families. Although sAD has a heterogeneous etiology and heritability of 70% to 80% (Gatz et al.,2006; Wingo et al., 2012), age is its most prominent biological risk factor (Carr et al., 1997) with APOE4 gene being an additional risk factor (Dorszewska et al., 2016). Female gender is also an important contributor that is partially explainable by the fact that postmenopausal women lose the protection that estrogens confer to neuronal mitochondria against beta-amyloid (AB) toxicity. Older women are also more likely than age-matched men to suffer from metabolic diseases, such as diabetes and obesity, that increase their chances of developing AD (Vina and Lloret 2010).
Most fAD patients have at least one affected first-degree relative (van Duijn et al., 1994; Campion et al., 1999; Jarmolowicz et al., 2014), and in 10% to 15% the mode of inheritance is autosomal dominant transmission (Campion et al., 1999; Jarmolowicz et al., 2014). fAD is triggered by gene mutations of amyloid precursor protein (APP) (chromosome 21), presenilin 1 (PSEN1) (chromosome 14), or presenilin 2 (PSEN2) (chromosome 1). This elicits AB aggregation in earlier years and the onset of the disease is as early as 20–30 years of age (Su et al., 2008; Muirhead et al., 2010) with the majority being diagnosed between 45 and 60 years.
Additional risk genes that have been identified by genome-wide association studies (GWAS) include: the gene for clusterin (CLU) also known as apolipoprotein J (localized on chromosome 8), the gene encoding the complement component (3b/4b) receptor 1 (CR1) (chromosome 1), the gene encoding PI-binding clathrin assembly protein (PICALM) (chromosome 11), the gene encoding the bridging integrator 1 (BIN1) (chromosome 2), and the disabled homolog 1 (DAB1) (chromosome 1). Additional novel risk loci associated with sAD are: sortilin-like receptor 1 (SORL1),triggering receptor expressed on myeloid cells 2 (TREM2),the membrane-spanning 4-domains, subfamily A (MS4A),ATP-binding cassette transporter A1 and A7 (ABCA1 and 7), methylenetetrahydrofolate dehydrogenase 1 (MTHFD1)and CD33 (Allen et al., 2012). These newly identified genes are involved in transport, lipid metabolism (Zhu et al., 2015;El gaamouch et al., 2016), immune response and APP metabolism (De Strooper and Karran, 2016).
The drugs currently approved by the US Food and Drug Administration (FDA) for AD treatment include cholinesterase inhibitors (CIs) such as galantamine which is indicated for mild to moderate AD and rivastigmine, donepezil for all stages of AD (Kim et al., 2017). Tacrine, a centrally acting anticholinesterase and indirect cholinergic agonist is less prescribed due to its hepatotoxicity. Memantine, a glutamate agonist is a therapeutic option for moderate to severe AD (McShane et al., 2006; Jan et al., 2017). While these drugs ameliorate symptoms in the early stages of AD, they show no evidence of stopping or reversing the neurodegenerative process (De la Monte, 2012). This could be because they do not directly target the underlying pathology in the degenerating neurons and they may be more effective if administered in the prodromal stages of the disease.
Of the 244 drugs for AD tested in clinical trials registered with clinicaltrials.gov, a National Institutes of Health (NIH)registry of publicly and privately funded clinical studies between 2002–2012, only one has successfully completed clinical trials and received approval from the FDA (https://www.alz.org/documents_custom/2017-facts-and-figures.pdf). So far, all clinical trials designed to lower levels of AB by either blocking activity of B or γ secretases, preventing AB aggregation, or promoting AB clearance by immunotherapy have failed (Cummings et al., 2014; Feldman et al., 2014) emphasizing an urgent need tofind new therapies for AD.
It is widely accepted that alterations in mitochondrial function and glucose metabolism are consistent antecedents leading to AD pathology including AB plaque and neurofi brillary tangles (Gibson and Shi, 2010). This mitochondrial dysfunction is characterized by impaired biogenesis and inefficient bioenergetics i.e. defects the activity of key respiratory enzymes and is accompanied by the generation and accumulation of reactive oxygen species (ROS) (Chen and Yan, 2010). As AB accumulates, it impairs cerebral blood flow (CBF). This leads to less glucose being available for energy expenditure continuing a vicious cycle that exacerbates the compromised CBF and resulting in the degeneration of neurons (Popa-Wagner et al., 2015).
Additionally, AB impaired electron transport chain (ETC)function promotes the phosphorylation and polymerization of tau, a mitochondrial protein involved in microtubule assembly. This causes generation of more ROS that further stimulates tau phosphorylation leading to neurofibrillary tangle formation and neurodegeneration (Simoncini et al., 2015).Post-mortem brains of AD patients have fewer mitochondria and increased presence of mitochondrial DNA (mtDNA) and mitochondrial proteins in the cytosol (Arun et al., 2016).
Numerous positron emission tomography (PET) studies demonstrate a decline in glucose utilization in the hippocampal and entorhinal cortical regions that precedes the clinical diagnosis of AD by decades and predict the cognitive decline in normal aging (Calsolaro and Edison, 2016) or the progression of patients from mild cognitive impairment(MCI) to AD (Chetelat et al., 2003) with high accuracy.Mitochondrial biogenesis is a highly regulated process that requires coordination and crosstalk between the nuclear and mitochondrial genomes and plays an essential role in maintaining an adequate functional neuronal mitochondrial mass by compensating for damaged mitochondria that have been eliminated (Onyango et al., 2016).
To achieve better outcomes in patients with AD, a paradigm that addresses the bioenergetic deficit in the vulnerable neurons of affected brain regions is needed. This can be achieved by targeting mitochondria and alleviating mitochondrial dysfunction in AD.
The Mitochondrion
A mitochondrion contains 2—10 copies of mtDNA (Reddy, 2008). The human mtDNA consists of a 16.5 kb, double-stranded, circular DNA molecule (Anderson et al.,1981). mtDNA contains 13 polypeptide genes that encode essential components of the ETC. mtDNA also encodes the 12S and 16S ribosomal RNA (rRNA) genes and the 22 transfer RNA (tRNA) genes required for mitochondrial protein synthesis (Reddy and Beal, 2005). Nuclear genes encode the remaining mitochondrial proteins, metabolic enzymes,DNA and RNA polymerases, ribosomal proteins, and mtDNA regulatory factors, such as mitochondrial transcription factor A. Nuclear mitochondrial proteins are synthesized in the cytoplasm and are subsequently transported into mitochondria. mtDNA is inherited exclusively from the mother and is present in thousands of copies per cell. Mitochondrial number and morphology are controlled by an equilibrium of mitochondrial fusion andfission (Chan, 2006) that is vital for metabolism, energy production, Ca2+signaling, ROS production, apoptosis, and senescence (Chen et al., 2005;McBride et al., 2006; Yu et al., 2006). Fusion allows the exchange of mitochondrial components including mtDNA between different mitochondria. mtDNA due to their proximity to the respiratory chain and a lack of protective histones have a very high mutation rate that is about ten times faster compared to the nuclear DNA (nDNA).
New mtDNA mutations arise frequently in the maternal lineage and initially present as a mixture of the wild-type and mutant mtDNAs, defining the so-called heteroplasmic state.mtDNA mutations are most often heteroplasmic (mixed population of normal and mutant mtDNAs). During cellular divisions, the mutant mtDNAs will be randomly segregated into the daughter’s cells and the percentage of mutant mtDNAs in different cell lineages will drift toward either pure mutant or normal (or homoplasmy) (Stewart and Chinnery,2015). As the percentage of mutant mtDNAs increases in the cell, energy output falls, resulting in an overall mitochondrial dysfunction in the cell. Hence, the ratio of mutant to normal mtDNA contributes to the severity of the disease. Severe mitochondrial damage impairs fusion resulting in fragmentation of mitochondria that are then selectively removed by an autophagic process called mitophagy (Kim et al., 2007).
Mitochondria are structurally and functionally altered in AD (Burte et al., 2015; Cai and Tammineni, 2016; Onyango et al., 2016), and compounds that are able to induce and/or restore their bioenergetic capacity present an attractive strategy AD therapy. Here we review nascent developments of mitochondrially targeted approaches that show promise for AD treatment.
Cellular Therapy
Cell-based therapies are a promising alternative currently being developed to enable the reversal of neurodegeneration in AD either directly by replacing injured neuron or indi-rectly by stimulating neuronal repairviaparacrine signaling at the injury site (Baraniak and McDevitt, 2010). Neurons and glial cells have successfully been generated from embryonic stem cells (ESCs), neural stem cells (NSCs), neural progenitor cells (NPCs), mesenchymal stem cells (MSCs), induced pluripotent stem cells (iPSCs), induced neuronal cells (iN),induced neuronal progenitor cells (iNPCs). Transplantation of NSCs into animal models of neurodegenerative diseases,including AD, increases the total amount of mtDNA, messenger RNA and protein levels of mitochondrial biogenesis-related factors as well as protein levels of mitochondrialfission genes. This results in a significant increase in the number of morphologically well-structured mitochondria in neurons and is associated with a reversal of cognitive defects, clinical improvement and life extension of these animals (Kim et al.,2013; Zhang et al., 2015; Mendivil-Perez et al., 2017). While still in its formative phase, this newfield shows great therapeutic promise for AD and other neurodegenerative diseases.
Drug Therapy
Targeting ROS
Targeting detrimental neuronal ROS at the production stage without affecting ROS signaling would be ideal in preventing and treating AD. In this regard, it has been shown that mitochondria-targeted antioxidants potently sequester reactive oxygen intermediates and confer greater protection against oxidative damage in the mitochondria than untargeted cellular antioxidants. The ability of mitochondria-targeted antioxidants to confer greater protection against oxidative damage in the mitochondria than untargeted cellular antioxidants provide has been attributed to their ability to cross the mitochondrial phospholipid bilayer and eliminate ROS where it is being generated (Oyewole and Birch-Machin, 2015).
These mitochondria-targeted antioxidants such as(10-(6′-plastoquinonyl) decyltriphenyl-phosphonium)(SkQ1), MitoQ, MitoTEMPOL and MitoVitE prevent apoptosis by mitigating the oxidative damage more effectively than untargeted antioxidants such as 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox) (Oyewole and Birch-Machin, 2015). Other such antioxidants include:4,5-dihydroxybenzene-1,3-disulfonate (Tiron), which has been engineered to accumulate within the mitochondria by permeabilizing the mitochondrial membrane (Fang et al.,2012) and astaxanthin, a mitochondrion-permeable antioxidant, that can penetrate the blood-brain barrier and is effective in preventing and treating macular degeneration(Piermarocchi et al., 2012; Wu et al., 2014).
Various compounds, such as coenzyme Q10(CoQ10), vitamin E, curcumin,Gingko biloba, melatonin and lipoic acid,have been demonstrated to reduce AB accumulation, protect mitochondria from AB toxicity, restore mitochondrial function and attenuate cognitive impairment in animal models of AD possess mitochondrial restoring and anti-oxidant properties (Du and Yan, 2010; Zhang et al., 2015).
Antioxidants can also be targeted to mitochondria through the use of small, aromatic-cationic sequence motif called Szeto-Schiller (SS) tetrapeptides which enables them to be delivered and localized to the inner mitochondrial membrane with an approximate 1,000–5,000-fold accumulation (Smith and Murphy, 2011; Jin et al., 2014). Novel XJB peptides which consist of an electron and ROS scavenger(4-NH2-TEMPO) conjugated to the Leo-D-Phe-Pro-Val-Orn fragment of gramicidin S have been invented. This pentapeptide fragment can specifically target the XJB peptides to mitochondria. One of these peptides, XJB-5-131, improved mitochondrial function and enhanced the survival of neurons in a mouse model of Huntington’s disease (Xun et al., 2012)and might offer a viable therapeutic opportunity in AD. Another approach of targeting mitochondria with small bioactive molecules involves polymer based nano-carriers. These include biodegradable poly-lactide-co-gylcolide (PLGA) like PLGA-CoQ10nanoparticles (Nehilla et al., 2008) although their biological effects are yet to be fully elucidated.
Targeting the inflammasome
Mitochondria are capable of regulating the pro-inflammatory response of the cell through activation of the inflammasome. The inflammasome is a multi-protein complex on which proIL-1B and proIL-18 processing occurs. The NLRP3 inflammasome, detects the inflammatory aggregates of AB and inactive IL-1B, and responds by secreting caspase-1(Casp-1) to activate IL-1B (Saco et al., 2014).
NLRP3 activation is crucial in the pathogenesis of AD(Walsh et al., 2014) and has been proposed to be associated with mitochondrial dysfunction including: mitochondrial ROS (Zhou et al., 2011), mitochondrion-derived damage associated molecular patterns (mtDAMPs), such as oxidized mitochondrial DNA (Shimada et al., 2012; Wilkins et al.,2015), and translocation of cardiolipin from the inner to the outer mitochondrial membrane (Iyer et al., 2013). Additionally, extracellular ATP at various concentrations can activate microglia and induce neuroprotective or neurotoxic effects by expressing pro- or anti-inflammatory cytokines (Inoue,2002; Davalos et al., 2005).
Several studies in cell lines, genetic rodent models, and humans indicate that redox control might serve as a bidirectional link between energy metabolism, redox control and neuroinflammatory responses in the brain that might serve as an integrated mechanism for AD etiology (Yin et al., 2016). It has been reported that small molecule inhibitors of the NLRP3 inflammasome ameliorate AD pathology in animal models of AD (Dempsey et al., 2017; Yin et al.,2017). Further, CAD-31, a safe, orally active and brain-penetrant neurotrophic drug that targets inflammation has been shown to reduce synaptic loss, normalize cognitive skills and enhance brain bioenergetics in genetic mouse models of AD(Daugherty et al., 2017).
Targeting the proteasome
The ubiquitin proteasome system (UPS) and mitochondria systems are tightly interdependent. Once a vicious cycle of dysfunction starts in diseases such as AD, it is difficult to identify which system was the trigger. Mitochondrial dysfunction and impairment of the UPS are two hallmarks of aging and both are implicated in AD (Riederer et al., 2011;Ross et al., 2015). Proteasome activation by small molecules is a promising strategy to treat or prevent neurodegenerative diseases characterized by the accumulation of toxic protein aggregates (Lee et al., 2010; Dantuma and Bott, 2014; Myeku et al., 2016). As a proof of concept, it has been shown that proteasome activation by genetic manipulation ameliorates the aging process and increases lifespan in different models includingC. elegans, human fibroblasts and yeast cells (Chondrogianni et al., 2015). Proteasome function is activated by a pathway involving protein kinase A and cyclic AMP (cAMP). Selective phosphodiesterase-4 inhibitors such as rolipram which increase cAMP levels have been shown to increase proteasome function, reduce aggregated tau levels,and improve cognitive performance and ameliorate the early stages of neurodegeneration in a genetically engineered mouse model of tauopathy (Myeku et al., 2016).
Proteosomal activity can also be enhanced by using Pyrazolone containing small molecules which block USP14,a proteasome-associated deubiquitinating enzyme that inhibits the processing of ubiquitin—protein complexes destined for degradation by the proteasome (Lee et al., 2010).PD169316 is a novel small molecule p38 MAPK inhibitor and a very potent activator of proteasome activity enhanced Proteolysis Targeting Chimeric (PROTAC)-mediated and ubiquitin-dependent protein degradation and decreases the levels of both overexpressed and endogenous α-synuclein in a bimolecular fluorescence complementation (BiFC) assay(Outeiro et al., 2008), without affecting the overall protein turnover. It also increased the viability of cells overexpressing toxic α-synuclein assemblies (Leestemaker et al., 2017).
Gene Therapy
Targeting mtDNA
mtDNA are relatively unstable and susceptible to damage because they lack histones and have limited enzymatic repair system, as well as their crucial role in oxidative phosphorylation (OXPHOS). As a result, mtDNA mutations accumulate with age (Smigrodzki and Khan, 2005; Casoli et al., 2015) and are a significant risk factor for AD (Swerdlow et al., 2014).
(i) The clustered regularly interspaced short palindromic repeats (CRISPR)/associated protein 9 (CRISPR/Cas9) technology has now been developed to produce mitochondrial sequence-specific cleavage with the potential of targeting specific mitochondrial genes (Jo et al., 2015). It utilizes a custom single guide RNA (sgRNA) fragment that acts as a guide tofind the piece of DNA to be modified and binds to it and recruits mitoCas9, whose localization is restricted to mitochondria matrix. This mitoCas9 could be applied to edit mtDNA together with gRNA expression vectors without affecting genomic DNA.
(ii) Transcription activator-like effector nucleases(TALENs) comprise a non-specific DNA-cleaving nuclease fused to a DNA-binding domain that can be easily engineered so that TALENs can target essentially any sequence.When directed specifically at the mtDNA, mitoTALENs(Bacman et al., 2013) can be used to cleave the mutated mtDNA, efficiently reducing the levels of the targeted pathogenic mtDNAs in the respective cell lines. This enables cells with heteroplasmic mutant mtDNA to recover respiratory capacity and oxidative phosphorylation enzymes activity(Hashimoto et al., 2015).
Targeting mitochondrial cholesterol
The accumulation of cholesterol in mitochondria can lead to mitochondrial dysfunction and may be a key step in AD progression. This dysfunction includes reduced fluidity of mitochondrial membranes (Colell et al., 2003), reduced ATP generation (Echegoyen et al., 1993; Yu et al., 2005) and decreased mitochondrial glutathione (GSH) import (Marí et al., 2006; Garcia-Ruiz et al., 2009) and may be a key step in AD progression (Aufschnaiter, et al., 2016). Furthermore,there is a direct link between altered membrane lipids and mitochondrial function, which is detrimental for brain bioenergetics (Rosales-Corral et al., 2012). Cholesterol turnover in the brain is modulated by cytochrome P450 46A1 (CYP46A1)which initiates the major pathway of its elimination. In the APP23 AD mouse model, AB peptides accumulate following inhibition of CYP46A1 expression resulting in widespread neuronal death compared to normal mice. On the other hand, decreasing CYP46A1 gene expression in hippocampal neurons of normal mice increases the cholesterol concentration in neurons with subsequent cognitive deficits and hippocampal atrophy due to apoptotic neuronal death (Djelti et al.,2015). Preclinical pharmacological tests are ongoing for gene therapy targeting CYP46A1 as a means to restore cholesterol metabolism in AD brain (Djelti et al., 2015) with clinical trials anticipated to begin in 2021 (www.brainvectis.com).
Biologics
The human mitochondrial genome can be manipulated from outside the cell to change expression and increase mitochondrial energy production. Mitochondrial transcription factor A (TFAM), has been engineered to rapidly pass through cell membranes and target mitochondria. Expression of human TFAM (hTFAM) significantly improved cognitive function,reducing accumulation of both 8-oxoguanine, an oxidized form of guanine, in mtDNA and intracellular AB in 3xTg-AD mice and increasing expression of transthyretin, known to inhibit AB aggregation (Oka et al., 2016). We previously showed that recombinant-human TFAM (rhTFAM) acts on cultured cells carrying a mtDNA disease (Iyer et al., 2012)as well as lab mice, energizing the DNA of the mice’s mitochondria, improving the memory of aged mice (Iyer et al.,2009; Thomas et al., 2012) and enabling them to run two times longer on their rotating rods than a control group cohort (Thomas et al., 2012).
Caloric Restriction (CR)
CR,i.e.the limitation of ingested calories without malnutrition, is known to enhance animal life span and prevent age-related diseases, including neurological deficits, brain atrophy, and cognitive decline (Colman et al., 2009). CR induces mitochondrial biogenesis (Cerqueira et al., 2012) in a NO·-mediated manner that culminates in increased mitophagy and the production of new, more efficient mitochondria that have reduced membrane potential, produce less ROS,consume increased levels of oxygen and exhibit an improved ATP/ROS ratio - leading to decreased energy expenditure(Onyango et al., 2010). In particular, the tissue-specific effects of CR include the prevention of the age-related loss of mtDNA in rat liver (Cassano et al., 2006) and the partial preservation of TFAM binding to mtDNA in rat brain with enhanced reserve respiratory capacity and improved survival in neurons (Picca et al., 2013).
Exercise
Endurance exercise (EE) is neuroprotective against AD.Exercise activates continuous oxidative stress that induces a series of counteractive mechanisms that enhance mitochondrial function and mitigate ROS-induced neurotoxicityi.e., mitohormesis (Onyango et al., 2010; Radak et al., 2016),and this is especially important in the hippocampus which is particularly sensitive to oxidative stress (Intlekofer and Cotman, 2013). In animal models of AD, physical exercise reduces the noxious effects of oxidative stress, the production of total cholesterol, and insulin resistance, while increasing vascularization and angiogenesis, improving glucose metabolism as well as neurotrophic functions, thereby facilitating neurogenesis and synaptogenesis, and as a consequence improving memory and cognitive functions (Paillard et al.,2015; Chen et al., 2016; Koo et al., 2016).
A combination of CR and EE is reported to deliver more beneficial effects than either regimen alone in ameliorating neurological and cognitive deficits (Cherif et al., 2016).
Conclusion
Mitochondrial impairment and loss play a critical role in neuronal degeneration and disease progression in AD.Damaged mitochondria are less bioenergetically efficient and produce increased amounts of ROS with detrimental structural and functional consequences for the AD neurons. Dysfunctional mitochondrial accumulate from the combination of impaired mitophagy, which can also induce injurious inflammatory responses, and inadequate neuronal mitochondrial biogenesis.
Identifying mechanisms that are critical in enhancing mitochondrial quality control, reducing bioenergetic defects while limiting generation of detrimental quantities of ROS may provide therapeutic opportunities that preserve neuronal viability and function and delay or reverse features of AD (Figure 1).
Author contributions:IGO wrote the manuscript and made thefigure.
Conflicts of interest:None declared.
Financial support:None.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
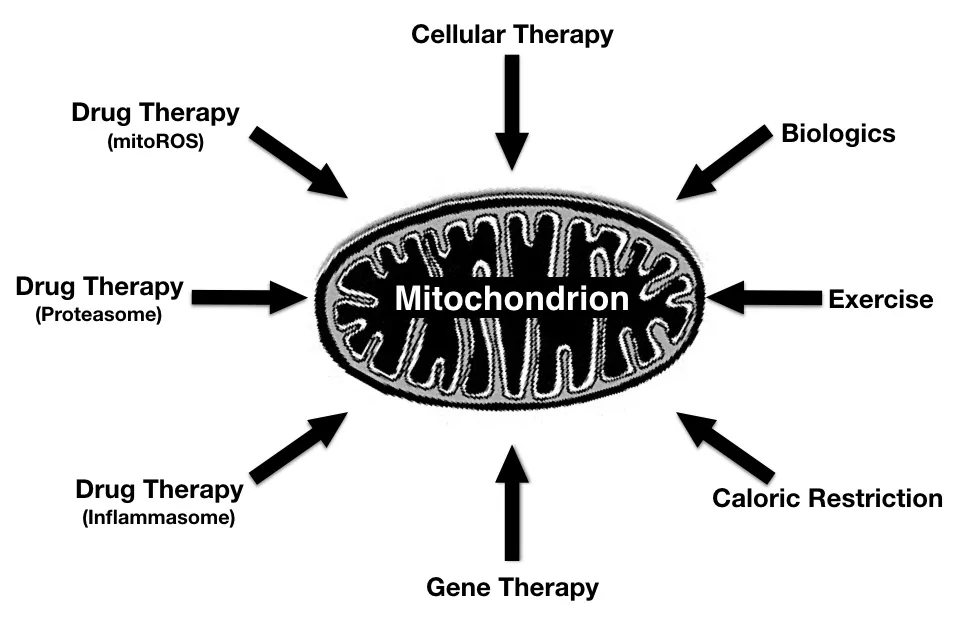
Figure 1 Approaches to enhance bioenergetic capacity in Alzheimer’s disease (AD).
Open access statement:This is an open access article distributed underthe terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under identical terms.
Open peer review report:
Reviewer:Aurel Popa-Wagner, University Medicine Rostock, Germany.
Comments to author:To date, all clinical trials designed to lower levels of AB by either blocking activity of B or γ secretases, preventing AB aggregation, or promoting AB clearance by immunotherapy have failed. The idea that alterations of mitochondrial function and glucose metabolism may lead to AD pathology is gaining acceptance in the scientific community. The authors make a survey of current therapies that might make mitochondria of AD patients bioenergetically more efficient. The article is well organized.
Allen M, Zou F, Chai HS, Younkin CS, Crook J, Pankratz VS, Carrasquillo MM, Rowley CN, Nair AA, Middha S, Maharjan S, Nguyen T,Ma L, Malphrus KG, Palusak R, Lincoln S, Bisceglio G, Georgescu C, Schultz D, Rakhshan F, et al. (2012) Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology 79:221-228.
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457-465.
Arun S, Liu L, Donmez G (2016) Mitochondrial biology and neurological diseases. Curr Neuropharmacol 14:143-154.
Aufschnaiter A, Kohler V, Diessl J, Peselj C, Carmona-Gutierrez D,KellerW, Buttner S (2016) Mitochondrial lipids in neurodegeneration. Cell Tissue Res 367:1-16.
Bacman SR, Williams SL, Pinto M, Peralta S, Moraes CT (2013) Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat Med 19:1111-1113.
Baraniak, PR, McDevitt, TC (2010) Stem cell paracrine actions and tissue regeneration. Regen Med 5:121-143.
Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P (2015) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11:11-24.
Cai Q, Tammineni P (2016) Alterations in mitochondrial quality control in Alzheimer’s disease. Front Cell Neurosci 10:24.
Calsolaro V, Edison P (2016) Alterations in glucose metabolism in Alzheimer’s disease. Recent Pat Endocr Metab Immune Drug Discov 10:31-39.
Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T (1999) Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet 65:664-670.
Carr DB, Goate A, Phil D, Morris JC (1997) Current concepts in the pathogenesis of Alzheimer’s disease. Am J Med 103:3S-10S.
Casoli T, Spazzafumo L, Di Stefano G, Conti F (2015) Role of diffuse low-level heteroplasmy of mitochondrial DNA in Alzheimer’s disease neurodegeneration. Front Aging Neurosci 7:pe142.
Cassano P, Sciancalepore AG, Lezza AM, Leeuwenburgh C, Cantatore P,Gadaleta MN (2006) Tissue-specific effect of age and caloric restriction diet on mitochondrial DNA content. Rejuvenation Res 9:211-214.
Cerqueira FM, Cunha FM, Laurindo FR, Kowaltowski AJ (2012) Calorie restriction increases cerebral mitochondrial respiratory capacity in a NO·-mediated mechanism: impact on neuronal survival. Free Radic Biol Med 52:123641.
Chan DC (2006) Mitochondrial fusion andfission in mammals. Ann Rev Cell Dev Biol 22:79-99.
Chen H, Chomyn A, Chan D (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280:26185-26192.
Chen JX, Yan SS (2010) Role of mitochondrial amyloid-beta in Alzheimer’s disease. J Alzheimers Dis. 20 Suppl 2:S569-578.
Chen W, Zhang X, Huang W (2016) Role of physical exercise in Alzheimer’s disease (Review). Biomed Rep 4:403-407.
Cherif A, Roelands B, Meeusen R, Chamari K (2016) Effects of intermittent fasting, caloric restriction, and ramadan intermittent fasting on cognitive performance at rest and during exercise in adults. Sports Med 46:35-47.
Chetelat G, Desgranges B, de la Sayette V, Viader F, Eustache F, Baron JC (2003) Mild cognitive impairment: can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology 60:1374-1377.
Chondrogianni N, Voutetakis K, Kapetanou M, Delitsikou V, Papaevgeniou N, Sakellari M, Lefaki M, Filippopoulou K, Gonos ES (2015) Proteasome activation: An innovative promising approach for delaying aging and retarding age-related diseases. Ageing Res Rev 23:37-55.
Colell A, Garcia-Ruiz C, Lluis JM, Coll O, Mari M, Fernandez-Checa JC(2003) Cholesterol impairs the adenine nucleotide translocator-mediated mitochondrial permeability transition through altered membrane fluidity. J Biol Chem 278:33928-33935.
Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, Anderson RM (2014) Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun 5:3557.
Cummings JL, Morstorf T, Zhong K (2014) Alzheimer’s disease drug-development pipe-line: few candidates, frequent failures. Alzheimers ResTher 6:37.
Dantuma NP, Bott LC (2014) The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution.Front Mol Neurosci 7:70.
Daugherty D, Goldberg J, Fischer W, Dargusch R, Maher P, Schubert D(2017) A novel Alzheimer’s disease drug candidate targeting inflammation and fatty acid metabolism. Alzheimers Res Ther 9:50.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR,Dustin ML, Gan WB (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8:752-758.
De la Monte SM (2012) Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res 9:35-66.
De Strooper B, Karran E (2016) The cellular phase of Alzheimer’s disease. Cell 164:603-615.
Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL,RobertsonAAB, Cooper MA, O’Neill LA, Lynch MA (2017) Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-B and cognitive function in APP/PS1 mice. Brain Behav Immun 61:306-316.
Djelti F, Braudeau J, Hudry E, Dhenain M, Varin J, Bieche I, Marquer C,Chali F, Ayciriex S, Auzeil N, Alves S, Langui D, Potier MC, Laprevote O, Vidaud M, Duyckaerts C, Miles R, Aubourg P, Cartier N (2015)CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer’s disease. Brain 138:2383-2398.
Dorszewska J, Prendecki M, Oczkowska A, Dezor M, Kozubski W (2016)Molecular basis of familial and sporadic alzheimer’s disease. Curr Alzheimer Res 13:952-963.
Du H, Yan SS (2010) Mitochondrial medicine for neurodegenerative diseases. Int. J. Biochem. Cell Biol 42:560-572.
Echegoyen S, Oliva EB, Sepulveda J, Diaz-Zagoya JC, Espinosa-Garcia MT, Pardo JP, Martinez F (1993) Cholesterol increase in mitochondria: its effect on inner-membrane functions, submitochondrial localization and ultrastructural morphology. Biochem J 289:703-708.
El Gaamouch F, Jing P, Xia J, Cai D (2016) Alzheimer’s disease risk genes and lipid regulators. J Alzheimers Dis 53:15-29.
Fang Y, Hu XH, Jia ZG, Xu MH, Guo ZY, Gao FH (2012) Tiron protects against UVB-induced senescence-like characteristics in human dermalfibroblasts by the inhibition of superoxide anion production and glutathione depletion. Australas J Dermatol 53:172-180.
Feldman HH, Haas M, Gandy S, Schoepp DD, Cross AJ, Mayeux R,Sperling RA, Fillit H, van de Hoef DL, Dougal S, Nye JS (2014) Alzheimer’s disease research and development: a call for a new research roadmap. Ann N Y Acad Sci 1313:1-16.
Garcia-Ruiz C, Mari M, Colell A, Morales A, Caballero F, Montero J,Terrones O, Basañez G, Fernández-Checa JC (2009) Mitochondrial cholesterol in health and disease. Histol Histopathol 24:117-132.
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S,Fiske A, Pedersen NL (2006) Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63:168-174.
Gibson GE, Shi Q (2010) A mitocentric view of Alzheimer’s disease suggests multi-faceted treatments. J Alzheimers Dis 20:S591-S607.
Hashimoto M, Bacman SR, Peralta S, Falk MJ, Chomyn A, Chan DC,Williams SL, and Moraes CT (2015) MitoTALEN: a general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol Ther 23:1592-1599.
Inoue K (2002) Microglial activation by purines and pyrimidines. Glia 40:156-163.
Intlekofer KA, Cotman CW (2013) Exercise counteracts declining hippocampal function in aging and Alzheimer’s disease. Neurobiol Dis 57:47-55.
Iyer S, Thomas R, Portell F, Dunham L, Quigley C, Bennett J Jr (2009)Recombinant mitochondrial transcription factor A with N-terminal mitochondrial transduction domain increases respiration and mitochondrial gene expression. Mitochondrion 9:196-203.
Iyer S, Bergquist K, Young K, Gnaiger E, Rao RR, Bennett JP Jr (2012)Mitochondrial gene therapy improves respiration, biogenesis, and transcription in G11778A Leber’s hereditary optic neuropathy and T8993G Leigh’s syndrome cells. Hum Gene Ther 23:647-657.
Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL,Sutterwala FS (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39:311-323.
Jan AT, Azam M, Rahman S, Almigeiti AMS, Choi DH, Lee EJ, Haq QMR, Choii(2017) Perspective insights into disease progression,diagnostics, and therapeutic approaches in Alzheimer’s disease: a judicious update. Front Aging Neurosci. 9:356.
Jarmolowicz AI, Chen HY, Panegyres PK (2014) The patterns of inheritance in early-onset dementia: Alzheimer’s disease and frontotemporal dementia. Am J Alzheimers Dis Other Demen 30:299-306.
Jin H, Kanthasamy A, Ghosh A, Anantharam V, Kalyanaraman B, Kanthasamy AG (2014) Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes. Biochim Biophys Acta 1842:1282-1294.
Jo A, Ham S, Lee GH, Lee YI, Kim S, Lee YS, Shin JH, Lee Y (2015) Effi-cient mitochondrial genome editing by CRISPR/Cas9. Biomed Res Int 2015:305716.
Kim I, Rodriguez-Enriquez S, Lemasters J (2007) Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462:245-253.
Kim SU, Lee HJ, Kim YB (2013) Neural stem cell-based treatment for neurodegenerative diseases. Neuropathology 33:491-504.
Kim SH, Kandiah N, Hsu JL, Suthisisang C, Udommongkol C, Dash A(2017) Beyond symptomatic effects: potential of donepezil as a neuroprotective agent and disease modifier in Alzheimer’s disease. Br J Pharmacol 174:4224-4232.
Koo JH, Kang EB, Kwon IS, Jang YC, Kim EJ, Lee Y, Cho IH, Cho JY(2015) Endurance exercise confers neuroprotective mitochondrial phenotypes in the brain of Alzheimer’s disease mice. FASEB J 29:S1055.35.
Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, Wilson SM, King RW, Finley D (2010) Enhancement of proteasome activity by a small-molecule inhibitor of USP14.Nature 467:179-184.
Leestemaker Y, de Jong A, Witting KF, Penning R, Schuurman K, Rodenko B, Zaal EA, van de Kooij B, Laufer S, Heck AJR, Borst J, Scheper W,Berkers CR, Ovaa H (2017) Proteasome activation by small molecules.Cell Chem Biol 24:725-736.
Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, García-Ruiz C (2006) Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis.Cell Metabolism 4:185-198.
McBride HM, Neuspiel M, Wasiak S (2006) Mitochondria: more than just a powerhouse Curr Biol 16:R551-R560.
McShane R, Areosa Sastre A, Minakaran N (2006) Memantine for dementia. Cochrane Database Syst Rev:CD003154.
Mendivil-Perez M, Soto-Mercado V, Guerra-Librero A, Fernandez-Gil BI, Florido J, Shen YQ, Tejada MA, Capilla-Gonzalez V, Rusanova I,Garcia-Verdugo JM, Acuña-Castroviejo D, López LC, Velez-Pardo C,Jimenez-Del-Rio M, Ferrer JM, Escames G (2017) Melatonin enhances neural stem cell differentiation and engraftment by increasing mitochondrial function. J Pineal Res 63.
Muirhead KE, Borger E, Aitken L, Conway SJ, Gunn-Moore FJ (2010) The consequences of mitochondrial amyloid betapeptide in Alzheimer’s disease. Biochem J 426:255-270.
Myeku N, Clelland CL, Emrani S, Kukushkin NV, Yu WH, Goldberg AL,Duff KE (2016) Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med 22:46-53.
Nehilla BJ, Bergkvist M, Popat KC, Desai TA (2008) Purified and surfactant-free coenzyme Q10-loaded biodegradable nanoparticles. Int J Pharm 348:107-114.
Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, Hyman BT, McLean PJ (2008) Formation of toxic oligomeric alpha- synuclein species in living cells. PLoS One 3:e1867.
Oka S, Leon J, Sakumi K, Ide T, Kang D, LaFerla FM, Nakabeppu Y (2016)Human mitochondrial transcriptional factor A breaks the mitochondria-mediated vicious cycle in Alzheimer’s disease. Sci Rep 6:37889.
Onyango IG, Lu J, Rodova M, Lezi E, Crafter AB, Swerdlow RH (2010)Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim Biophys Acta 1802:228-234.
Onyango IG, Dennis J, Khan SM (2016) Mitochondrial dysfunction in Alzheimer’s disease and the rationale for bioenergetics based therapies.Aging Dis 7:201-214.
Oyewole AO, Birch-Machin MA (2015) Mitochondria-targeted antioxidants. FASEB J 29:4766-4771.
Paillard T, Rolland Y, de Souto Barreto P (2015) Protective effects of physical exercise in Alzheimer’s disease and Parkinson’s disease: a narrative review. J Clin Neurol 11:212-219.
Picca A, Fracasso F, Pesce V, Cantatore P, Joseph AM, Leeuwenburgh C, Gadaleta MN, Lezza AM (2013) Age- and calorie restriction-related changes in rat brain mitochondrial DNA and TFAM binding. Age 35:1607-1620.
Piermarocchi S, Saviano S, Parisi V, Tedeschi M, Panozzo G, Scarpa G,Boschi G, Lo Giudice G, Carmis Study Group (2012) Carotenoids in Age-related Maculopathy Italian Study (CARMIS): two-year results of a randomized study. Eur J Ophthalmol 22:216-225.
Popa-Wagner A, Buga AM, Popescu B, Muresanu D (2015) Vascular cognitive impairment, dementia, aging and energy demand. A vicious cycle.J Neural Transm 122:S47-S54.
Radak Z, Suzuki K, Higuchi M, Balogh L, Boldogh I, Koltai E (2016) Physical exercise, reactive oxygen species and neuroprotection. Free Radic Biol Med 98:187-196.
Reddy PH (2008) Mitochondrial medicine for aging and neurodegenerative diseases. Neuromol Med 10:291-315.
Reddy PH, Beal MF (2005) Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res Brain Res Rev 49:618-632.
Riederer BM, Leuba G, Vernay A, Riederer IM (2011) The role of the ubiquitin proteasome system in Alzheimer’s disease. Exp Biol Med 236:268-276.
Rosales-Corral SA, Lopez-Armas G, Cruz-Ramos J, Melnikov VG, Tan DX,Manchester LC, Munoz R, Reiter RJ (2012) Alterations in lipid levels of mito-chondrial membranes induced by amyloid-beta: A protective role of melatonin. Int J Alzheimers Dis 2012:459806.
Ross JM, Olson L, Coppotelli G (2015) Mitochondrial and ubiquitin proteasome system dysfunction in ageing and disease: two sides of the same coin? Int J Mol Sci 16:19458-19476.
Saco T, Parthasarathy PT, Cho Y, Lockey RF, Kolliputi N (2014) Inflammasome: a new trigger of Alzheimer’s disease. Frontiers in Aging Neuroscience 6:80.
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, Rentsendorj A, Vargas M,Guerrero C, Wang Y, Fitzgerald KA, Underhill DM, Town T, Arditi M (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36:401-414.
Simoncini C, Orsucci D, Caldarazzo E, Siciliano G, Bonuccelli U, Mancuso M (2015) Alzheimer’s pathogenesis and its link to the mitochondrion.Oxid Med Cell Longev 2015:803942.
Smigrodzki RM, Khan SM (2005) Mitochondrial microheteroplasmy and a theory of aging and age-related disease. Rejuvenation Res 8:172-198.
Smith RA, Murphy MP (2011) Mitochondria-targeted antioxidants as therapies. Discov Med 11:106-114.
Stewart JB, Chinnery PF (2015) The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet 16:530-142.
Su B, Wang X, Nunomura A, Moreira PI, Lee HG, Perry G, Smith MA,Zhu X (2008) Oxidative stress signaling in Alzheimer’s disease. Curr Alzheimer Res 5:525-532.
Swerdlow RH, Burns JM, Khan SM (2014) The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta 1842:1219-1231.
Thinakaran G (1999) The role of presenilins in Alzheimer’s disease. J Clin Invest 104:1321-1327.
Thomas RR, Khan SM, Smigrodzki RM, Onyango IG, Dennis J, Khan OM,Portell FR, Bennett JP (2012) RhTFAM treatment stimulates mitochondrial oxidative metabolism and improves memory in aged mice. Aging 4:620-635.
van Duijn CM, de Knijff P, Cruts M, Wehnert A, Havekes LM, Hofman A, Van Broeckhoven C (1994) Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer’s disease. Nat Genet 7:74-78.
Vina J, Lloret A (2010) Why women have more Alzheimer’s disease than men: gender and mitochondrial toxicity of amyloid-beta peptide. J Alzheimers Dis. 20:S527-533.
Walsh JG, Reinke SN, Mamik MK, McKenzie BA, Maingat F, Branton WG,Broadhurst DI, Power C (2014) Rapid in flammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology 11:35.
Wilkins HM, Carl SM, Weber SG, Ramanujan SA, Festoff BW, Linseman DA, Swerdlow RH (2015) Mitochondrial lysates induce inflammation and Alzheimer’s disease-relevant changes in microglial and neuronal cells. J Alzheimers Dis 45:305-318.
Wingo TS, Lah JJ, Levey AI, Cutler D (2012) Autosomal recessive causes likely in early-onset Alzheimer disease. Arch Neurol 69:59-64.
Wu W, Wang X, Xiang Q, Meng X, Peng Y, Du N, Liu Z, Sun Q, Wang C,Liu X (2014) Astaxanthin alleviates brain aging in rats by attenuating oxidative stress and increasing BDNF levels. Food Funct 5:158-166.
Xun Z, Rivera-Sanchez S, Ayala-Pena S, Lim J, Budworth H, Skoda EM,Robbins PD, Niedernhofer LJ, Wipf P, McMurray CT (2012) Targeting of XJB-5-131 to mitochondria suppresses oxidative DNA damage and motor decline in a mouse model of Huntington’s disease. Cell Rep 2:1137-1142.
Yin J, Zhao F, Chojnacki JE, Fulp J, Klein WL, Zhang S, Zhu X (2017)NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer’s disease. Mol Neurobiol doi: 10.1007/s12035-017-0467-9.
Yin F, Sancheti H, Patil I, Cadenas E (2016) Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic Biol Med 100:108-122.
Yu W, Gong JS, Ko M, Garver WS, Yanagisawa K, Michikawa M (2005)Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J Biol Chem 280:11731-11739.
Yu T, Robotham JL, Yoon Y (2006) Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A 103:2653-2658.
Zhang W, Gu GJ, Shen X, Zhang Q, Wang GM, Wang PJ (2015) Neural stem cell transplantation enhances mitochondrial biogenesis in a transgenic mouse model of Alzheimer’s disease-like pathology. Neurobiol Aging 36:1282-1292.
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 in flammasome activation. Nature 469:221-225.
Zhu L, Zhong M, Elder GA, Sano M, Holtzman DM, Gandy S, Cardozo C,Haroutunian V, Robakis NK, Cai D (2015) Phospholipid dysregulation contributes to ApoE4-associated cognitive deficits in Alzheimer’s disease pathogenesis. Proc Natl Acad Sci U S A 112:11965-11970.

- 中国神经再生研究(英文版)的其它文章
- Neural Regeneration Research: Information for Authors
- Conundrums and confusions regarding how polyethylene glycol-fusion produces excellent behavioral recovery after peripheral nerve injuries
- LETTER FROM THE EDITORS-IN-CHIEF
- Brain injury and neural stem cells
- Tackling dipeptidyl peptidase IV in neurological disorders
- Stem cells for spinal cord injuries bearing translational potential