
阴离子对Acidithiobacillus ferrooxidans氧化活性及次生铁矿物形成影响
2018-03-02 05:24宋永伟王鹤茹曹艳晓周立祥中南财经政法大学环境科学与工程系湖北武汉430073南京农业大学环境工程系江苏南京0095
中国环境科学 2018年2期
宋永伟,陈 婷,王鹤茹,杨 俊,曹艳晓,周立祥 (.中南财经政法大学环境科学与工程系,湖北 武汉430073;.南京农业大学环境工程系,江苏 南京 0095)
AMD中存在的嗜酸性氧化亚铁硫杆菌(A.ferrooxidans)能够高效催化Fe2+向Fe3+转化,并伴随着Fe3+水解产生施氏矿物、黄铁矾等次生铁矿物[9-12].已经证实,上述矿物对重(类)金属离子有较大的吸附或共沉淀作用,是较为理想的吸附材料[13-19].由上启示,生物成因次生铁矿物不仅可以有效去除Fe离子,还可吸持去除有毒有害元素,从而减轻环境的污染.促使AMD中Fe离子向次生铁矿物转变具有一定的科学研究价值.
A.ferrooxidans介导的生物矿化法受微生物活性影响,反应条件较为温和,因此生物成因次生铁矿物形成的因素也很多,如反应时间、反应温度、Fe2+浓度、阳离子种类/浓度、晶种种类等[20-25].次生铁矿物生物合成的实质是Fe2+→Fe3+→Fe8O8(OH)6SO4或MFe3(SO4)2(OH)6的Fe2+氧化和Fe3+水解过程,氧化产物Fe3+作为过渡离子,其供应速率决定了次生铁矿物的合成行为.可见,A.ferrooxidans的Fe2+氧化能力间接制约着次生铁矿物的形成过程,其与A.ferrooxidans活性密切相关.影响A.ferrooxidans活性的因素包括pH值、温度、O2/CO2浓度、金属离子等[26-31].此外,过量阴离子对A.ferrooxidans氧化活性也具有一定的抑制作用.已有报道称,浓度为10mmol/L的Cl-能显著延缓A.ferrooxidans的生长;在超过94mmol/L时能导致A.ferrooxidans死亡;当或浓度大于300mmol/L时,则完全抑制A.ferrooxidans生长[32-33].上述研究结果比较是基于不同菌株或能源底物而言,但关于阴离子对A.ferrooxidansFe2+氧化活性及其介导次生铁矿物形成的综合影响鲜见报道.那么,受上述3种阴离子影响后,生物成因次生铁矿物的相对成矿能力(总Fe沉淀率)和次生铁矿物矿相之间的差异问题均还需进一步研究和考证.
本研究以去除酸性矿山废水中Fe离子及重(类)金属元素为目的,通过A.ferrooxidans介导的次生铁矿物生物矿化法,研究酸性环境下Cl-、、浓度对溶液中Fe2+氧化率、总Fe沉淀率、次生铁矿物矿相的影响.研究结果可为促使酸性硫酸盐体系中Fe向次生铁矿物的转变和调控提供必要的理论依据.
1 材料与方法
1.1 供试材料
改良9K液体培养基:(NH4)2SO43.5g、KCl 0.119g、K2HPO40.058g、Ca(NO3)24H2O 0.0168g、MgSO47H2O 0.583g,蒸馏水1000mL, pH=2.50,121℃灭菌30min.
A.ferrooxidans休止细胞:将A.ferrooxidans接种在改良9K培养基中,置于28℃、180r/min摇床(QYC-2102C)中振荡培养,待指数生长阶段后期停止培养(约2~3d).随后将培养液经定性滤纸过滤以除去生成的次生铁矿物,将滤液以10000×g的相对离心力(TGL205,4℃,10min)离心收集菌体,并用pH=1.50的酸水(H2SO4配制)清洗3次,以除去各种杂离子.将这些菌体悬浮于pH=2.50的酸水(H2SO4配制),所得即为A.ferrooxidans浓缩菌液[34].
1.2 试验设置
在系列含有若干去离子水的500mL三角瓶中,按7.84g/L (即140mmol/L)的Fe2+浓度加入FeSO47H2O,用1:1的H2SO4调上述所有体系pH值至2.50,然后分别采用KCl、KNO3、K3PO4作为3种阴离子来源进行调节,设置Cl-、、浓度梯度分别为0,25,100,250,500,750mmol/L.随后接种A.ferrooxidans休止细胞悬浮液,并补充少量去离子水,使体系的有效容积为250mL,A.ferrooxidans密度约为5×107cells/mL.将上述三角瓶置于28℃、180r/min摇床中振荡培养168h.培养过程中,定期取液体样约1mL过0.22μm滤膜(取样前使三角瓶预先静置5min,使次生矿物完全沉降,然后取上清液),测定和计算Fe2+浓度、总Fe沉淀率的变化情况.培养期间采用称重法定时补加因蒸发减少的水分.培养终点时,用中速定性滤纸收集合成的次生矿物,用去离子水清洗2次以去除杂质,60℃烘干后称重并进行矿物相鉴定.不同处理均设置3个重复.
1.3 测定方法
采用pHS-3C精密pH计测定溶液pH值;Fe2+和总Fe浓度采用邻啡罗啉比色法测定;矿物相采用X射线衍射仪测定(XRD,Bruker D8),测试工作条件为:管电压40kV,管电流40mA,扫描区间10°~80°(2θ),步长0.01°,扫描速率6°/min,Cu靶.
1.4 数据处理与统计分析
实验数据采用SPSS(SPSS 21for windows)统计软件进行统计分析,分差分析后采用SNK(Student-Newman-Keuls test)方法进行多重比较.
2 结果
2.1 3种阴离子对体系pH值、Fe2+的影响

图1 不同浓度阴离子处理下体系pH值的变化情况Fig.1 Change of pH value with different concentrations of Cl-, , and
本试验使用的A.ferrooxidans是休止细胞,不含有供细胞生长的营养元素,细胞不会发生增殖,但仍具有较强的生物催化氧化能力.图1~2显示的是当溶液Fe2+浓度为7.84g/L时,不同阴离子对酸性硫酸盐体系pH值和Fe2+浓度的影响情况.

图2 不同浓度阴离子处理下体系Fe2+的变化情况Fig.2 Change of Fe2+ with different concentrations of Cl-, and
A. ferrooxidans氧化Fe2+成矿包含两步酸效应:一是耗酸的Fe2+生物氧化过程,二是产酸的Fe3+水解成矿过程.因此,通过反应体系pH值的变化趋势即可判断A. ferrooxidans的氧化能力.由图1知,当初始pH=2.50时,Cl-和对体系pH值基本没有影响,而浓度则表现出一定的缓冲性能,pH值在0h时出现波动.这可能与是多元弱酸离子有关,pH=2.50时主要以H3PO4和型体存在.培养至24h时,Cl-≤25、≤100、≤100mmol/L范围内,各处理pH值均上升至2.70左右,并随着培养时间延长开始逐渐下降,说明Fe2+生物氧化(耗酸)和Fe3+水解成矿(产酸)过程能够正常进行,该阴离子浓度水平对A.ferrooxidans的催化氧化活性没有产生抑制作用.相反,低浓度阴离子反而有助于体系pH值下降,表现在pH值相对下降速度更快和下降程度更低.以为例,当培养时间分别为96h和168h时,0、25、100mmol/L的所对应的pH值分别为2.30、2.24、1.98和1.99、1.85、1.75.从阴离子种类来看,适当浓度更易加速体系pH值在短时间内下降.同为25mmol/L的Cl-、NO3、在培养至96h时,溶液pH值分别为2.22、2.24、1.94.当Cl-=100、=250mmol/L时,A. ferrooxidans氧化活性受到一定的冲击,但并没有完全失活,后期能够恢复氧化功能,表现为pH值分别在96、120h时出现回升和缓慢下降的趋势.通过阴离子对体系pH值的变化可以判断,A.ferrooxidans对3种阴离子的耐受性依次为>>Cl-.由图2可知,当阴离子浓度在A.ferrooxidans耐受范围内时,其对Fe2+的生物氧化过程基本没有影响,体系中7.84g/L的Fe2+均在72~96h内被氧化完全,这与pH值变化趋势存在一定差异(图1).根据A.ferrooxidans介导的生物成矿酸效应理论可以推断,低浓度阴离子体系pH值的下降速度主要受Fe3+水解成矿过程影响.从Fe2+氧化速率来看,A.ferrooxidans对能源物质Fe2+的氧化利用主要集中在24~72h,期间Fe2+累积氧化率>80%.在0~24h内,Fe2+氧化率偏低可能与高速离心导致A. ferrooxidans受损有关,使得其缓冲期延长.当Cl-≥250mmol/L、≥250mmol/L、≥500mmol/L时,A.ferrooxidans则完全抑制失活,溶液中Fe2+浓度在反应前后基本没有变化.可见,阴离子离子种类及浓度对Fe2+的生物氧化具有重要影响,主要体现在高浓度离子水平对A.ferrooxidans氧化能力的抑制作用.郭勤[35]等研究表明,当Cl-浓度大于85mmol/L,细菌生长就完全受到抑制.张成桂[32]等以单质硫作为底物考察硫杆菌活性时,发现Cl-、、对细胞氧化能力完全抑制的临界浓度分别为50、50、300mmol/L.前人结果与本研究相差较大,可能受供试菌株种类不同的影响.相对于Cl-和A.ferrooxidans对具有较强耐受性的原因在于是细胞内核酸、磷脂及ATP的必需组分,能够参与ATP和ADP的形成,在能量积累和转换及生理代谢等发面扮演重要的角色.
2.2 3种阴离子对体系Fe3+水解成矿的影响

图3 不同浓度阴离子处理下体系总Fe的变化情况Fig.3 Change of total Fe with different concentrations of Cl-, , and
图3描述了总Fe沉淀率随培养时间的变化趋势.其实质是Fe2+氧化和Fe3+水解形成次生铁矿物而从液相转移到固相的过程[36].受A.ferrooxidans活性影响,反应初期Fe2+氧化速度较慢而减缓Fe3+水解成矿,各处理总Fe沉淀主要集中在24~120h,该时间段内体系具备较大的Fe3+供应速率,使次生铁矿物形成推动力发生改变,在一定程度上促进矿物的加速形成[37-39].适当提高阴离子浓度有利于可溶性Fe的生物成矿去除,各阴离子的贡献能力依次为>>Cl-.以阴离子浓度25mmol/L为例,添加Cl-、PO43-体系反应终点的总Fe累积沉淀率分别为43.67%、50.67%、58.04%,与对照处理相比分别提高了7.31%、14.31%、21.68%.
然而,由图2可知,低浓度水平的各阴离子对Fe2+生物氧化过程并没有显著影响,即水解成矿所需Fe3+供应速率基本一致.由此可以推断,各体系中总Fe沉淀率存在明显差异并非由阴离子种类或浓度直接决定.结合次生铁矿物合成过程可知,当酸性环境体系中存在一价阳离子时,会参与黄铁矾的合成过程,一价阳离子浓度越高,越有利于反应的正向进行,消耗单位Fe3+所合成纯黄铁矾质量约为纯施氏矿物的1.5倍以上.而本研究中阴离子均以K盐的形式添加,可能改变了次生铁矿物的形成途径,使合成过程更倾向于黄铁矾,从而提高总铁沉淀率(由图5佐证).此外,当阴离子投加浓度相同时,K3PO4中K+浓度为KCl、KNO3的3倍,理论上合成的次生铁矿物量应高得多,而从图4来看,当阴离子浓度为25mmol/L时,Cl-、、收集矿物质量分别为2.98、3.25、3.46g,并非呈倍数递增关系.猜测跟次生铁矿物形成过程的产H+速度有关,持续水解成矿过程以致体系pH值过低可能导致次生铁矿物易于扩散和溶解[40-41].结合图1可以发现,当反应时间在96h时,25mmol/L的体系pH值就下降至2.0以下,而相同浓度的Cl-、则需要144h才能达到该酸度水平.刘奋武[42]等在酸性环境下考察过施氏矿物的稳定性,结果表明,施氏矿物在pH=3.0体系中振荡72h后溶解率只有3.34%,而相同时间内pH=2.0体系中的溶解率则高达61.46%.

图4 不同浓度阴离子处理下生物成因次生铁矿物质量的变化情况Fig.4 Change of biogenic secondary iron mineral quality with different concentrations of Cl-, , and
2.3 3种阴离子对体系次生铁矿物相的影响
图5为阴离子浓度在0~100mmol/L时收集次生铁矿物XRD图谱.前已述及,浓度在A.ferrooxidans耐受范围内时,次生铁矿物的合成过程主要受一价阳离子K+浓度的影响,而高浓度阴离子则会通过抑制A.ferrooxidans的Fe2+生物氧化活性,从而间接阻碍Fe3+的水解成矿过程.
参考JCPDS晶型黄钾铁矾(No: 22-0827)和非晶型施氏矿物(No: 47-1775)的标准XRD图谱可知[43],图5中对照处理(0mmol/L)为FeSO4-A.ferrooxidans-H2O体系,根据衍射峰位置及相对强度(2θ=35.16°),分析生成矿物为无定型的施氏矿物[44].当阴离子浓度提高至25,100mmol/L时,除Cl-=100mmol/L处理外,FeSO4-A.ferrooxidans-K+-H2O体系获得次生铁矿物均为纯净的黄钾铁矾[45],且随着阴离子浓度提高特征衍射峰愈加明显.Bai等[46]研究结果表明,在Fe2+初始浓度为160mmol/L溶液中,当K+≥16.0mmol/L(即Fe2+/K+≤10)时形成的次生铁矿物均为单一黄钾铁矾.而本试验中Fe2+/K+分别为5.6、1.9,与前人研究结果基本一致.因100mmol/L浓度Cl-对A.ferrooxidans氧化活性的抑制作用,导致Fe3+供应速度不足,虽然成矾导向离子K+浓度满足条件,但仍不利于反应向合成黄钾铁矾的方向进行,收集的矿物为施氏矿物和黄钾铁矾的混合物.
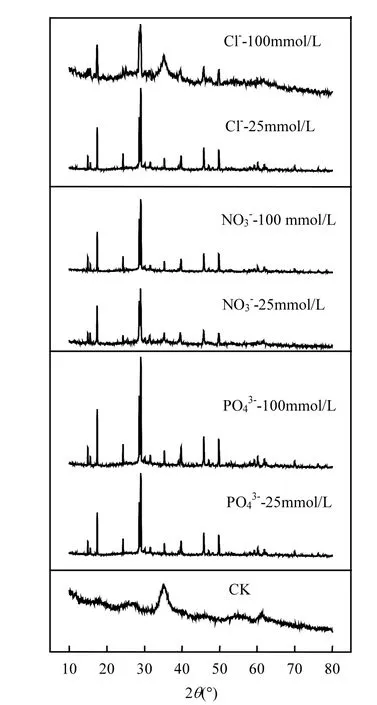
图5 生物成因次生铁矿物的XRD图谱Fig.5 XRD patterns of biogenic secondary iron minerals
3 结论
3.1 高浓度阴离子对A.ferrooxidans氧化Fe2+能力具有一定的抑制作用.A.ferrooxidans对阴离子的耐受性依次为>>Cl-.
3.2 在A.ferrooxidans耐受范围内时,阴离子对Fe2+的生物氧化基本没有影响, 140mmol/L的Fe2+均在72~96h内被氧化完全.
3.3 高浓度阴离子会通过抑制A.ferrooxidans氧化活性,从而间接影响Fe3+的水解成矿过程,导致体系总Fe沉淀率降低和次生铁矿物产量减少.
[1] Singer P C, Stumm W. Acidic mine drainage: the ratedetermining step [J]. Science, 1970,167(3921):1121-1123.
[2] Vhahangwele M. A novel technology for neutralizing acidity and attenuating toxic chemical species from acid mine drainage using cryptocrystalline magnesite tailings [J]. Journal of Water Process Engineering, 2016,10(6):67-77.
[3] Wu Z L, Zou L C, Chen J H, et al. Column bioleaching characteristic of copper and iron from Zijinshan sulfide ores by acid mine drainage [J]. International Journal of Mineral Processing, 2016,149:18-24.
[4] Wei X, Wolfe F A. Minerals and Mine Drainage [J]. Water Environment Research, 2013,85(10):1515-1547.
[5] Liu G W, Bai R C. Development of the acidic mining wastewater treatment technology [J]. Applied Mechanics and Materials,2013,295-298:1372-1375.
[6] Song Y W, Wang M, Liang J R, et al. High-rate precipitation of iron as jarosite by using a combination process of electrolytic reduction and biological oxidation [J]. Hydrometallurgy,2014,143(3):23-27.
[7] Lee W C, Lee S W, Yun S T, et al. A novel method of utilizing permeable reactive kiddle (PRK) for the remediation of acid mine drainage [J]. Journal of Hazardous Materials, 2016,301:332-341.
[8] Meschke K, Herdegen V, Aubel T, et al. Treatment of opencast lignite mining induced acid mine drainage (AMD) using a rotating microfiltration system [J]. Journal of Environmental Chemical Engineering, 2015,4(4):2848-2856.
[9] Diao Z H, Shi T H, Wang S Z, et al. Silane-based coatings on the pyrite for remediation of acid mine drainage [J]. Water Research,2013,47(13):4391-4402.
[10] Valente T, Grande J A, Torre M L, et al. Mineralogy and environmental relevance of AMD-precipitates from the Tharsis mines, Iberian Pyrite Belt (SW, Spain) [J]. Applied Geochemistry,2013,39(8):11-25.
[11] Zhu J Y, Gan M, Zhang D, et al. The nature of Schwertmannite and Jarosite mediated by two strains of Acidithiobacillus ferrooxidans with different ferrous oxidation ability [J]. Materials Science and Engineering C, 2013,33(5):2679-2685.
[12] 宋永伟,赵博文,霍敏波,等.温度对嗜酸性硫杆菌活性和生物成因次生铁矿物形成的影响 [J]. 环境科学, 2013,34(8):3264-3271.
[13] Zhang S L, Jia S Y, Yu B, et al. Sulfidization of As(V)-containing schwertmannite and its impact on arsenic mobilization [J].Chemical Geology, 2016,420(20):270-279.
[14] Gan M, Sun S G, Zheng Z H, et al. Adsorption of Cr(VI) and Cu(II) by AlPO4modified biosynthetic schwertmannite [J].Applied Surface Science, 2015,356(30):986-997.
[15] Mihone K M, Hana F, Sanda R, et al. Assessment of metal risks from different depths of jarosite tailing waste of Trepça Zinc Industry, Kosovo based on BCR procedure [J]. Journal of Geochemical Exploration, 2015,148:161-168.
[16] 廖岳华.施氏矿物的生物合成及去除水中砷的效果与机理研究[D]. 南京:南京农业大学, 2008.
[17] 陈福星,周立祥.生物催化合成的施氏矿物对废水中Cr(VI)的吸附 [J]. 中国环境科学, 2006,26(1):11-15.
[18] 王长秋,马生凤,鲁安怀.黄钾铁矾类矿物沉淀去除Cr(VI)的初步研究 [J]. 矿物岩石地球化学通报, 2006,25(4):335-338.
[19] Asta M P, Cama J, Martínez M, et al. Arsenic removal by goethite and jarosite in acidic conditions and its environmental implications [J]. Journal of Hazardous Materials, 2009,171(1-3):965-972.
[20] 邓志明,周正华.湿法炼锌浸出沉铁探讨 [J]. 湖南有色金属,2002,18(1):23-25,45.
[21] Dutrizac J E, Kaiman S. Synthesis and properties of jarosite-type compounds [J]. Canadian Mineralogist, 1976,14:151-158.
[22] Dutrizac J E. The effectiveness of jarosite species for precipitating sodium jarosite [J]. Journal of the Minerals, Metals and Materials Society, 1999,51(12):30-32.
[23] 宋永伟,王鹤茹,梁剑茹,等.温度和pH对生物成因羟基硫酸铁矿物的综合影响研究 [J]. 环境科学学报, 2016,36(10):3683-3690.
[24] 刘奋武,高诗颖,王 敏,等.镁离子对氧化亚铁硫杆菌生物合成次生铁矿物的影响 [J]. 中国环境科学, 2014,34(3):713-719.
[25] 刘奋武,高诗颖,崔春红,等.Ca2+对酸性硫酸盐环境中次生铁矿物合成的影响 [J]. 中国环境科学, 2015,35(4):1142-1148.
[26] Nemati M, Harrison S T L, Hansford G S, et al. Biological oxidation of ferrous sulphate byThiobacillus ferrooxidans: a review on the kinetic aspects [J]. Biochemical Engineering Journal,1998,3(1):171-190.
[27] 宋永伟,王鹤茹,曹艳晓,等.低分子有机酸对硫杆菌活性的抑制作用及对土壤重金属脱除的影响 [J]. 环境科学, 2016,37(5):368-375.
[28] Mousavi S M, Yaghmaei S, Salimi F, et al. Influence of process variables on biooxidation of ferrous sulfate by an indigenousAcidithiobacillus ferrooxidans. Part Ⅱ: Bioreactor experiments[J]. Fuel, 2007,86(7/8):993-999.
[29] Kumar S R, Gandhi K S. Modelling of Fe2+oxidation byThiobacillus ferrooxidans[J]. Applied Microbiology and Biotechnology, 1990,33(5):524-528.
[30] 刘 清,徐伟昌,张 宇.重金属离子对氧化亚铁硫杆菌活性的影响 [J]. 铀矿冶, 2004,23(3):155-157.
[31] 刘欣伟,冯雅丽,李浩然,等.镁离子浓度对氧化亚铁硫杆菌生长动力学的影响 [J]. 中国有色金属学报, 2012,26(8):2353-2359.[32] 张成桂,张 倩,王 晶,等.阴离子对嗜酸性氧化亚铁硫杆菌生长和硫氧化活性的影响 [J]. 中国有色金属学报, 2009,19(12):2237-2242.
[33] Harahuc L, Lizama H M, Suzuki I. Selective inhibition of the oxidation of Ferrous iron or sulfur inThiobacillus ferrooxidans[J].Applied and Environmental Microbiology, 2000,66(3):1031-1037.
[34] Liao Y, Zhou L, Liang J, et al. Biosynthesis of schwertmannite byAcidithiobacillus ferrooxidanscell suspensions under different pH condition [J]. Materials Science and Engineering C, 2009,29(1):211-215.
[35] 郭 勤,韩文艳,李 江,等.氯离子对氧化亚铁硫杆菌和氧化亚铁钩端微螺菌活性的影响 [J]. 有色金属, 2015,(1):42-45.
[36] Gramp J P, Sandy Jones F, Bigham J M, et al. Monovalent cation concentrations determine the types of Fe(III) hydroxysulfate precipitates formed in bioleach solutions [J]. Hydrometallurgy,2008,94(1-4):29-33.
[37] 柏双友.酸性富铁硫酸盐环境中生物成因次生羟基硫酸铁矿物形成及其机理 [D]. 南京:南京农业大学, 2012.
[38] Sasaki K, Konno H. Morphology of jarosite-group compounds precipitated from biologically and chemically oxidized Fe ions [J].The Canadian mineralogist, 2000,38(1):45-56.
[39] Regenspurg S, Brand A, Peiffer S. Formation and stability of schwertmannite in acid mining lakes [J]. Geochimica Et Cosmochimica Acta, 2004,68(6):1185-1197.
[40] Dold B. Dissolution kinetics of schwertmannite and ferrihydrite in oxidized mine samples and their detection by differential X-ray diffraction (DXRD) [J]. Applied Geochemistry, 2003,18(10):1531-1540.
[41] Loan M, Richmond W R, Parkinson G M. On the crystal growth of nanoscale schwertmannite [J]. Journal of Crystal Growth,2005,275(1/2):1875-1881.
[42] 刘奋武,卜玉山,田国举.温度与pH对生物合成施氏矿物在酸性环境中溶解行为及对Cu2+吸附效果的影响 [J]. 环境科学学报,2013,33(9):2445-2451.
[43] JCPDS (Joint Committee on Powder Diffraction Standards).Mineral Powder Diffraction Files [Z]. International Center for Diffraction Data, Swarthmore: Pennsyvania, 2002.
[44] Eskandarpour A, Onyango M S, Ochieng A, et al. Removal of fluoride ions from aqueous solution at low p H using schwertmannite [J].Journal of Hazardous Materials, 2008,152(2):571-519.
[45] Wang H M, Bigham J M, Tuovinen O H. Formation of schwertmannite and its transformation to jarosite in the presence of acidophilic iron-oxidizing microorganisms [J]. Materials Science and Engineering C, 2006,26(4):588-592.
[46] Bai S Y, Xu Z H, Wang M, et al. Both initial concentrations of Fe(II) and monovalent cations jointly determine the formation of biogenic iron hydroxysulfate precipitates in acidic sulfate-rich environments [J]. Materials Sciences and Engineering C, 2012,32(8):2323-2329.
猜你喜欢
中国钢铁业(2022年1期)2022-05-11
中国钢铁业(2022年2期)2022-05-11
矿产勘查(2021年3期)2021-07-20
石油沥青(2021年1期)2021-04-13
矿产勘查(2020年2期)2020-12-28
矿产勘查(2020年6期)2020-12-25
矿产勘查(2020年6期)2020-12-25
昆钢科技(2020年6期)2020-03-29
中国钢铁业(2019年10期)2019-06-11
中国洗涤用品工业(2015年8期)2015-02-28
