
Non-drip Filtration Plates for High Performance LC-MS/MSAnalysis of 5 Phenothiazine-class Antipsychotics in Blood
2018-03-01 02:49WANGZhaohongZHAOMengCHUJianxinJIANGWenhuiLIHongLIUYong
刑事技术 2018年1期
WANG Zhaohong, ZHAO Meng, CHU Jianxin, JIANG Wenhui, LI Hong, LIU Yong
(1. Procuratoral Technology and Information Research Center, Supreme People’s Procuratorate, Beijing 100041, China;2. People’s Procuratorate of Zhejiang Province, Hangzhou 310000, China;3. Hangzhou Municipal People’s Procuratorate, Hangzhou 310000, China;4. Key Laboratory of Narcotics Assay and Control Technology, Ministry of Public Security, Kunming 650000, China)
1 Introduction
Chlorpromazine (CPZ), promethazine (PMZ), tri fl uoperazine (TPZ), perphenazine (PPZ) and chlorprothixene(CPT) belong to the group of phenothiazine antipsychotics since all of them are derivatives of phenothiazine with different chains attached at the 2- or 10- position[1]. These compounds are usually prescribed for controlling manic symptoms, reducing or eliminating psychotic episodes such as hallucination, delusions and anxiety[2]. Due to their sedative and hypnotic effects, phenothiazines are readily used by criminals to engage into illegal acts. Numerous cases of fatal overdose have also involved the use of multiple antipsychotic drugs[3]. In such a situation, it is imperative to establish a robust method to determine phenothiazine drugs for both forensic toxicology and clinical therapeutic drug monitoring.
According to reports, phenothiazine drugs are mainly metabolized in the body and their concentrations in blood are usually very low. Therefore, a variety of analytical methods have been established to detect trace amounts of phenothiazine drugs in bio- fl uids or other biological samples, aiming to improve their sensitivity of detection.GC[4]and LC[5-7], combined with various detectors and LC-MS/MS[10-12], are probably the most popular alternatives. Some researchers are also focusing on sample preparation and try to achieve high analyte recoveries to reach higher sensitivity. For example, dispersive liquidliquid micro-extraction[8]and one-pot extraction[9]were introduced into usage. Unfortunately, the sample preparation by these methods is cumbersome in design and time-consuming.
Captiva ND Lipids (Non-drip) is a non-drip 96-well filtration plate, simple to use, specially designed to effectively remove proteins and phospholipids from biofluids. Used with Captiva ND Lipids, blood extracts are almost free from the endogenous matrix interferers including lipids, proteins and surfactants. Therefore, ion suppression in LC-MS/MS is signi fi cantly reduced, leading to enhanced sensitivity and precision for trace analysis. In our study, Captiva ND Lipids was combined with UPLC-MS/MS for rapid analysis of 5 phenothiazineclass antipsychotics in human blood. Results of full validation demonstrated that the method possesses favorable features at the items of perfect selectivity and speci fi city,high detection sensitivity, improved peak shapes, reproducible retention time and high sample throughput. The proposed method provides an appealing alternative for the forensic toxicology laboratory as well as for therapeutic drug monitoring in hospital.
2 The experimental
2.1 Chemicals and reagents
CPZ, PMZ, TPZ, PPZ, CPT and chloropromazine-d3(CPZ-d3) were purchased from the National Institute for the Control of Pharmaceutical and Biological Products.All reagents were HPLC grade. Acetonitrile and methanol were purchased from Fisher Scienti fi c (Fairlawn, NJ).Formic acid was obtained from Sigma-Aldrich (USA).Deionized water was generated with an Elix water puri fication system (Millipore Corp., Molscheim, France).
2.2 Apparatus
Analyses were performed on a Waters ACQUITY UPLC system with the Xevo TQ-S MS as the detector.Chromatographic data were recorded and analyzed by MasslynxTMsoftware (Version 4.1).
2.3 UPLC conditions
Separations were carried out on an ACQUITY UPLC HSS SB C18 column (2.1 mm x 50 mm, 1.7 μm;Waters, USA), being maintained at the temperature of 35°C and a constant fl ow rate of 0.5 mL/min. The mobile phase was composed of acetonitrile (A) and 0.1 % formic acid (B) in puri fi ed water. The gradient program was selected as shown in Table 1. The sample injection volume was 5 μL.

Table 1 Gradient program of the established method
2.4 MS/MS conditions
The quantitative analysis of 5 analytes was performed in the MRM mode. Positive ionization mode (ESI+) was used for all analytes. The MS parameters were: capillary voltage 0.5 kV, desolvation gas temperature 400 °C, desolvation gas fl ow 800 L/h, cone gas fl ow 150 L/h and colli-sion gas fl ow 0.16 mL/min. Each analyte was identi fi ed by two characteristic MRM transitions as shown in Table 2.
2.5 Blood samples
Blank human whole blood samples were obtained from volunteers with informed consent. The samples were stored at -20 °C. The project was approved by the Ethics Committee on Human Research of the Faculty of Health Science of Ministry of Health. Standard stock solutions of each analyte (2 mg/mL) were prepared in methanol. A mixed standard solution containing the 5 analytes at 1 μg/mL of each was prepared from the standard stock solutions. Spiked blood samples were prepared by the mixed standard stock solution adding into blank blood at appropriate concentrations in the range of 0.2-20 ng/mL.

Table 2 The two resulting MRM transitions per analyte and corresponding settings for UPLC-ESI-MS/MS system
Captiva ND Lipids (Varian, USA) plates were used for blood sample preparation. Aliquots of 100 μL spiked blood sample (with 1 ng/mL CPZ-d3as internal Standard, IS) were transferred into a 96-well fi ltration plate.Then, 400 μL of 0.1 % formic acid in methanol was added to each sample. Subsequently, the 96-well filtration plate was placed on a vacuum manifold and fi ltered under 20 psi pressure to remove any precipitated proteins.The filtrate was collected into the 96-well receiving plate. Thereafter, the 96-well receiving plate was capped and placed in the sample manager and 5 μL of each fi ltrate was used for UPLC-MS/MS analysis.
3 Results and discussion
3.1 Selectivity and speci fi city
Selectivity and speci fi city were evaluated by analyzing six different sources of blank blood samples, spiked blood samples and neat standard solutions (1 ng/mL),examining whether there were any interferences between the matrix signals and those from the analytes and the deuterated standard. A representative chromatogram was given in Fig. 1, showing that all the analytes of interest were free of any interference from the matrix and separated on the baseline. Conventional protein precipitation of blood samples usually leaves major interferences from the matrix. However, in samples prepared with Captiva ND Lipids under identical precipitation conditions,over 97% of phosphatidylcholines, lysophospholipids included, are removed. This lipid depletion signi fi cantly reduces the interferences from the compounds by the method itself.
3.2 Linearity and sensitivity
Calibration curves were prepared with seven concentration points (0.2, 0.5, 1.0, 2.0, 5.0, 10.0 and 20.0 ng/mL) for each drug in blood. Calibration was performed by linear regression of peak-area ratios of the drugs to the internal standard versus the respective standard concentration. The deuterated internal standards remained at a concentration of 1 ng/mL. Method sensitivity was systematically evaluated with a signal-to-noise ratio of at least 3. The regression equations, correlation coef fi cients and LODs for each analyte are listed in Table 3. All of the calibration curves were linear in the range of 0.2~20 ng/mL with correlation coef fi cients (r2) higher than 0.999.The LODs were 3-10 pg/mL and were far less than those reported in the literature (Table 6). When blood samples being prepared with Captiva ND Lipids, the relative responses were higher than the conventional protein precipitation. This increased sensitivity results from a significant reduction in ion suppression since Captiva ND Lipids virtually removes lipids, proteins and surfactants.

Table 3 Regression equations, correlation coef fi cients and LODs for the analytes in blood
3.3 Precision and accuracy
Measurements of precision and accuracy were carried out based on three concentration levels: 0.2, 2.0 and 20.0 ng/mL. Intra-/inter-day precisions were evaluated by analyzing fi ve replicates of each level for three consecutive days. Precision was measured as the relative standard deviation (RSD). Accuracy was calculated according to the following formula: accuracy (%) = [(calculated value- nominal value)/nominal value] ×100. From the data listed in Table 4, it can be seen that the accuracy was within -2 to 2 % and the RSDs were below 10 %. With Captiva ND Lipids, the integrity of the peak shapes was maintained. The smoother aspect of the peak made integration easier and more precise, and the reproducibility of quantitation also being improved.

Fig.1 Quantitative ion chromatograms of each analyte in neat standard solution (A), blank blood sample (B) and spiking blood sample (C)

Table 4 Accuracy, intra- and inter-day precision data (values given in %) of the UPLC-ESI-MS/MS assay for each analyte (n=5)
3.4 Extraction ef fi ciency and matrix effects
The extraction ef fi ciency was estimated at three levels with fi ve replicates at each concentration. Extraction recovery was calculated by comparison of the peak area obtained when the analytes and the IS were added before fi ltration (n=5), with those obtained when both the analytes and IS were added after the fi ltration step (n=5).
Matrix effects were determined by measuring five replicates of neat standard at three concentration levels and the filtrates from five spiked blank blood samples,and were calculated as the percentage of peak area in the presence of matrix relative to the peak area without matrix. Data are listed in Table 5. The extraction recoveries of samples containing the analyts at three concentration levels were 73.9 % - 103.3 % and the results of matrix effects were also satisfactory. The ion suppression effects from non-removed phosphatidylcholine interferences are obvious in a post-column infusion experiment with albuterol. When using Captiva NDLipids, the cleanliness of the extract is signi fi cantly improved compared to conventional protein precipitation. Consequently, ion suppression is dramatically reduced with no noticeable loss in MS response.

Table 5 Extraction ef fi ciency and matrix effects (values given as %) of the UPLC-ESI-MS/MS assay for each analyte at low,medium and high concentrations (ng/mL, n=5)
3.5 Sample stability
Blood samples were prepared of containing the analytes at three different concentrations (n=5 at each level)and kept at a temperature of 18 °C for 24 h and in one freezer at −20 °C for a month to evaluate the short- and long-term stability. To evaluate the stability of processed blood samples, fi ltrates of simulated blood samples containing the analytes at three different concentrations (n=5 at each level) were kept for 24 h in an autosampler at 10°C as well as in a freezer at −20 °C. For the evaluation of the stability after three freeze-thaw cycles, spiked blood samples were frozen at −20 °C for 24 h. After this period, the samples were again thawed and frozen for 24 h and this process was repeated until the third thawing cycle when the samples were processed and analyzed.All the stability-evaluating samples were determined by freshly-prepared calibration curves and compared with the reference concentrations. It was found that all measurements were within 15 % of the nominal concentrations, indicating that the samples were stable in each of the above storage conditions.
3.6 Comparison with reported methods
A comparison of the validation results for the proposed method with those of methods reported in the literature is given in Table 6. The established method has a wide range of analytes, tiny sample volume, time-saving for sample preparation, and much lower detection limit compared to the reported methods. These satisfactory results should be attributed to Captiva ND Lipids which uses a simple, streamlined and easy-to-follow 3-step process, making the quanti fi cation for trace analysis more precise and reproducible, reducing costs and increasing productivity. The described method was especially suitable for rapid analysis of trace analytes in small volume of specimens.
3.7 Application to forensic case samples
The established method was applied to several real forensic cases. Bloods from six fatalities were collected at autopsy and analyzed by the proposed method after appropriate dilution. Case details and analytical results are shown in Table 7. CPZ, PMZ and CPT had been each detected in two different cases, respectively. The proposed method was proved to be a robust and fast alternative in forensic toxicology analysis.
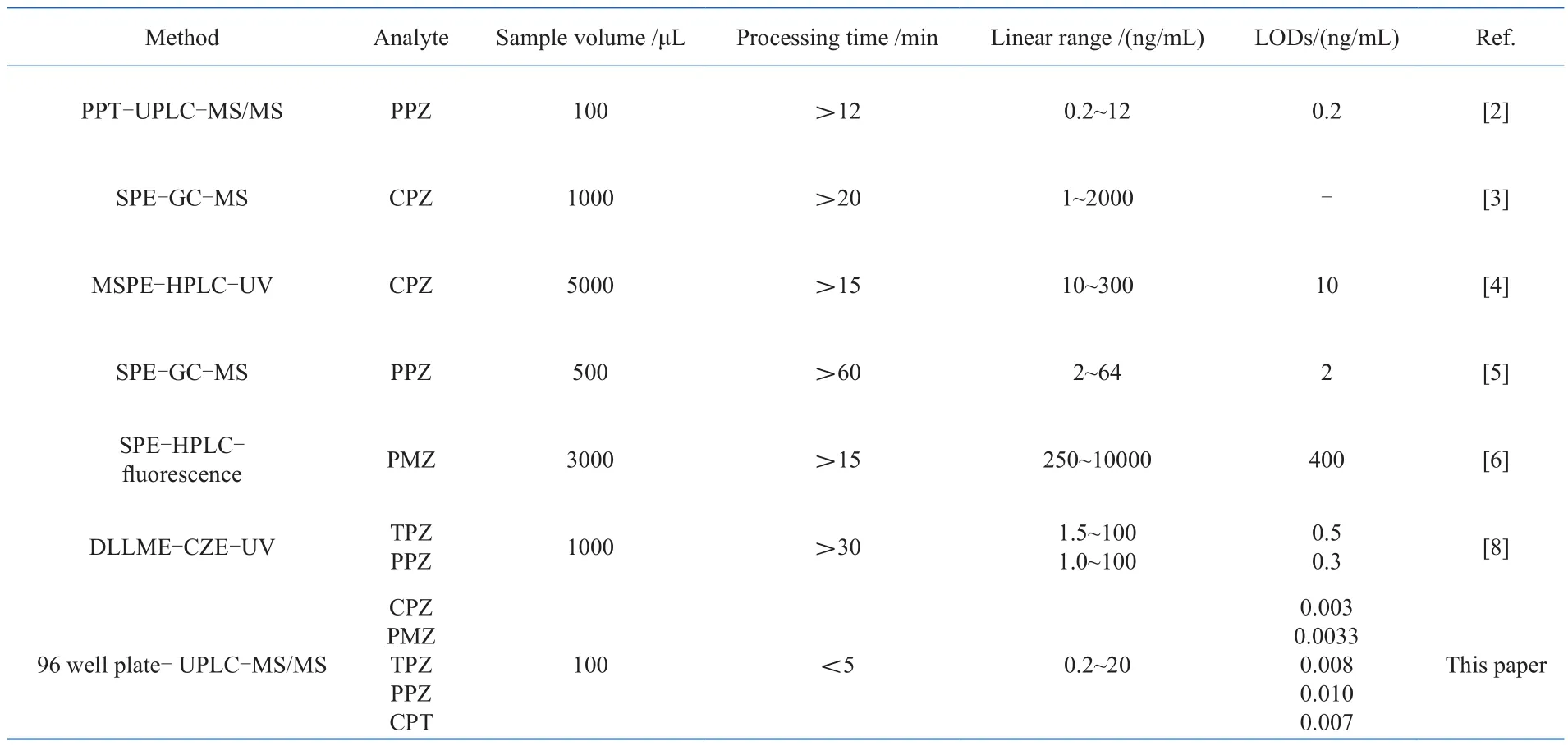
Table 6 Comparison of the proposed method with reported choices

Table 7 Details of forensic cases and detection results using the established method
4 Conclusion
In the present work, 5 phenothiazine-class antipsychotics were measured in small-volume human blood samples by UPLC-MS/MS with quite simple sample preparation. This method has many advantages including high-throughput, high sensitivity, simple sample preparation and short analysis time. The proposed method has been successfully applied in several fatal cases and the entirely satisfactory results were obtained. With these highly practical and desirable features, Captiva ND Lipids combining with UPLC-MS/MS method constitutes a simpler alternative for the rapid analysis of trace amounts of phenothiazine-class antipsychotic drugs for the forensic toxicology laboratory as well as for therapeutic drug monitoring in hospital.
[1] XU L, LI L, HUANG J, et al. Analysis of perphenazine and fluphenazine by capillary electrophoresis coupled with tris(2,2’-bipyridyl) ruthenium (II) electrochemiluminescence detection[J]. Talanta, 2014, 118: 1-6.
[2] JUENKE J M, MCGRAW J P, MCMILLIN G A, et al. Performance characteristics and patient comparison of the ARK Diagnostics levetiracetam immunoassay with an ultra-high performance liquid chromatography with tandem mass spectrometry detection method[J]. Clinica Chimica Acta, 2012, 18: 529-531.
[3] SASAKI C, SHINOZUKA T, MURAKAMI C, et al. Simultaneous determination of 5 psychotropic drugs of various types in an autopsy case of acute multiple drug poisoning[J]. Forensic Science International. 2013, 227:1-3.
[4] TURUNEN E, LEHTONEN M, JARVINEN T, et al. Development and validation of a gas chromatographic-mass spectrometric method for quantitative determination of perphenazine in rabbit plasma after sublingual administration[J]. Journal of Chromatography B ,2008, 872: 1-2.
[5] PONDER G W, STEWART J T. A liquid chromatographic method for the determination of promethazine enantiomers in human urine and serum using solid-phase extraction and fl uorescence detection[J]. Journal of Pharmaceutical & Biomedical Analysis, 1995, 13: 9.
[6] FOGLIA J P, SORISIO D, KIRSHNER M A, et al. Quantitative determination of perphenazine and its metabolites in plasma by high-performance liquid chromatography and coulometric detection[J]. Journal of Chromatography B Biomedical Applications, 1995, 668: 291-297.
[7] PINTO MA, SOUZA I D, Queiroz M E. Determination of drugs in plasma samples by disposable pipette extraction with C18-BSA phase and liquid chromatography-tandem mass spectrometry[J]. Journal of Pharmaceutical & Biomedical Analysis, 2017, 139: 116-124.
[8] DZIOMBA S, KOWALSKI P, SLOMINSKA A, et al. Field-ampli fi ed sample injection coupled with pseudo-isotachophoresis technique for sensitive determination of selected psychiatric drugs in human urine samples after dispersive liquid-liquid microextraction[J]. Analytica Chimica Acta, 2014, 811: 88-93.
[9] MATSUTA S, NAKANISHI K, MIKI A, et al. Development of a simple one-pot extraction method for various drugs and metabolites of forensic interest in blood by modifying the QuECh-ERS method[J]. Forensic Science International, 2013, 232: 1-3.
[10] DOMINGUES D S, PINTO M A, SOUZA I D, et al. Determination of drugs in plasma samples by high-performance liquid chromatography-tandem mass spectrometry for therapeutic drug monitoring of schizophrenic patients[J]. Journal of Analytical Toxicology, 2016, 40(1): 28-36.
[11] CAI H L, DENG Y, FANG P F, et al. A sensitive LC-MS/MS method for analysis of pericyazine in presence of 7-hydroxypericyazine and pericyazine sulphoxide in human plasma and its application to a comparative bioequivalence study in Chinese healthy volunteers[J]. Journal of Pharmaceutical &Biomedical Analysis, 2017, 20(135):67-74.
[12] UENKE J M, BROWN P I, UNRY F M, et al. Simultaneous UPLC-MS/MS assay for the detection of the traditional antipsychotics haloperidol, fluphenazine, perphenazine,and thiothixene in serum and plasma[J]. Clinica Chimica Acta., 2013,423:32-34.
