
杂多酸对Co-Pd/TiO2催化剂催化CH4-CO2两步梯阶转化合成乙醇和乙酸的影响
2018-02-20 03:23:00时培祥陶诗琪高东若
天然气化工—C1化学与化工 2018年6期
宋 刚,时培祥,陶诗琪,高东若,黄 伟
(太原理工大学煤化工研究所,煤科学与技术教育部和山西省重点实验室,山西 太原 030024)
能源和环境问题是当今世界所面临的两大课题。当前,以煤炭、石油作为主要燃料的国家,已面临严重的环境污染,加上化石燃料有限储量减少的双重危机日益加深,严重影响了经济社会的可持续发展。这就决定了我们不仅要重视节能,而且更重要的是要开发能源资源高效利用的新途径,从而减少温室气体排放,保护人类赖以生存的环境。
天然气作为一种优质、高效、清洁的化石燃料,在自然界中的蕴藏是十分丰富的,其储量远远超过石油,已经被广泛地应用于国民生活和生产的各个领域。随着不可再生资源的日益枯竭和天然气资源开发利用技术的不断提高,21世纪已经成为以天然气为主的能源时代。同时,天然气化工经过几十年的努力和发展已具有较为成熟的工艺过程[1-2]:天然气先转化成合成气,再制成煤油、柴油等液体燃料或者合成氨、甲醇等具有高附加值的化学品,甲醇可再进一步羰基化合成更为重要的有机化工原料乙酸等。但是以上的工艺过程比较复杂,条件苛刻,需要大量的能量输入。
CO2作为大气组成的一部分,在自然界中的储量十分丰富,在国民经济各部门中均有着十分广泛的用途。但是,随着现代化工业社会的不断进步与发展,过多的燃烧煤炭、石油和天然气,大量排放汽车尾气,使大量的CO2气体进入大气造成了严重的温室效应,对国民经济和自然环境有着巨大危害。基于这一现状,许多科研团队正在积极探索CO2转化的新路径,中科院大连化物所孙剑、葛庆杰研究员[3]发现了CO2高效转化的新过程,通过设计一种新型多功能复合催化剂,首次实现了CO2直接加氢制取高辛烷值汽油,这样不仅可有效降低CO2造成的温室效应,还可减轻对传统化石能源的依赖。但是CO2分子非常稳定难以活化,与费托合成[4]路线相比,CO2与氢分子的催化反应更易生成甲烷、甲醇、甲酸等小分子化合物,很难生成长链的液态烃燃料。
基于对能源高效利用和环境保护政策地积极响应,本课题组提出将储量巨大的CH4资源与化石能源利用排放量最大的污染物CO2相结合,探索出一条非氧、低能耗的CH4-CO2转化路线,其核心思想是将反应拆分成两步,有效地绕过了直接合成乙酸的热力学限制[5-13],实现了原子经济的高效利用,具有重要理论科学意义及应用价值。
根据课题组前期研究结果:在CH4-CO2两步梯阶转化直接合成乙醇和乙酸的实验过程中,Co-Pd/TiO2是最优良的催化剂,但载体TiO2存在比表面积小,表面酸性较弱等不足。针对这一问题,李志勤等[11]研究了无机酸改性对催化剂催化活性和选择性的影响,结果表明,酸处理改性改善了催化剂表面酸性,提高了催化剂活化CH4的能力。结合该反应机理可以发现:CH4在催化剂表面的解离吸附和氧化偶联是提高产物收率的关键性步骤。而固体杂多酸可以同时解决以上问题。
杂多酸是一类由氧原子桥接金属原子形成的金属-氧簇化合物。杂多酸具有良好的催化性能,是高效的双功能催化剂,即有酸催化性能,又具有氧化还原催化性能。王德胜等[14]归纳总结了杂多酸的分类及其主要应用,它可用作以芳烃烷基化和脱烷基反应、酯化反应、脱水/化合反应、氧化还原反应以及开环、缩合、加成和醚化反应等。中科院兰州化物所[15]研究了杂多酸改性催化剂对甲烷氧化偶联反应性能的影响,结果表明,改性的Na-W-Mn/SiO2系列催化剂反应性能突出的原因是W与Na形成了对反应有利的Na-O-W活性中心。基于上述分析结果,本文选择用磷钨酸对溶胶凝胶法制备的Co-Pd/TiO2催化剂进行改性,制备了不同浓度磷钨酸改性的Co-Pd/TiO2催化剂,在自制的双管固定床步阶反应器[16]上研究了其催化性能,并关联了其结构特性。
1 实验部分
1.1 试剂
异丙醇钛,AR,上海阿拉丁生化科技股份有限公司;Co(NO3)2·6H2O,AR,上海阿拉丁生化科技股份有限公司;PdCl2,AR,上海麦克林生化科技有限公司;无水乙醇,AR,天津市科密欧化学试剂有限公司;盐酸,36%~38%,太化集团公司化工三厂;冰醋酸,AR,天津市科密欧化学试剂有限公司;磷钨酸,AR,天津市光复精细化工研究所;CH4、H2、N2和 CO2纯度均为99.99%,太原钢铁公司。
1.2 催化剂的制备
采用溶胶凝胶法制备Co-Pd/TiO2催化剂。首先,将计量的异丙醇钛溶于含有一定量冰醋酸的无水乙醇中,记为溶液A;然后,按金属负载量称取一定量的PdCl2和Co(NO3)2·6H2O分别溶于盐酸和乙醇中,搅拌一段时间使其彻底溶解后加入到剧烈搅拌的A溶液中,调节pH≈1,搅拌1h后用蠕动泵定速缓慢滴加(12滴/min)适量的蒸馏水,继续搅拌1h后放置于室温下老化一段时间,在110℃空气中干燥3h,500℃空气中焙烧2h,制得Co-Pd/TiO2催化剂(其中 Co的质量分数为 7%,Pd的质量分数为3.5%)。
采用过量浸渍法制备磷钨酸改性双金属催化剂。首先配制等体积不同浓度的磷钨酸溶液,将上述制备的Co-Pd/TiO2催化剂等质量分别加入到不同浓度的磷钨酸溶液中,搅拌过夜,用蒸馏水多次洗涤后于110℃空气中干燥3h,500℃空气中焙烧2h,制得磷钨酸改性的Co-Pd/TiO2催化剂,压片、造粒,取40~60目备用;磷钨酸溶液的浓度分别为0.00,0.02,0.04,0.06 mol/L, 改性催化剂分别记为CAT0、CAT2、CAT4、CAT6。
1.3 催化活性的评价
催化剂的评价是在自主设计的连续式步阶反应器上进行的,反应器是由两个内径(8mm)相同的反应管组成,每个反应管中催化剂的加入量均为1.5g,均装填在以石英砂(40~70目)为床层的中心恒温区。催化剂在还原气氛下(30%H2和70%N2)程序升温至400℃还原2h,在N2气氛下降温至150℃进行反应,CH4和CO2中分别混有一定量的H2和H2O(g)(氢气和水蒸汽有利于目标产物的生成与脱附),CH4首先进入反应器A进行活化,同时CO2进入反应器B,当A反应器反应结束后,电磁阀换向,此时CO2进入反应器A与活化的CH4进行插入反应,CH4进入反应器B继续活化,当一次反应结束后,电磁阀再次换向,如此重复循环。反应产物通过冷凝器液化处理收集,手动液相进样用气相色谱GC-950氢焰检测分析。
1.4 催化剂表征
XRD表征采用日本生产的RigakuD/max-2500型粉末X-射线衍射仪,Ni滤片,Cu靶,Kα辐射,管电压40kV,电流30mA,连续扫描法,扫描宽度2θ为 10°~80°,扫描速度 8°/min。
NH3-TPD-MS表征采用TP-5000吸附仪进行,催化剂用量为100mg,在400℃的H2/N2混合气氛下预处理,50℃恒温脉冲NH3吸附至饱和,吹扫后,程序升温至750℃进行脱附,TCD检测并记录脱附曲线。
XPS表征采用ESCALAB250型光电子能谱仪检测,半球状电子分析仪,激光源为单色器Al靶(hv=1486.6eV),以 C1(Eb=284.6eV)校正核电效应。
BET表征采用美国生产的Quantachrome SI系列吸附仪于-196℃进行N2吸附测定。测定前将催化剂在200℃真空脱气3h,吸附质为液氮,根据BET测定催化剂的比表面积,采用BJH方法得到孔径分布。
FT-IR表征采用NICOLET360型红外光谱仪进行,测试时,将少量催化剂与干燥的溴化钾按1∶100的质量比混合,研磨后压片对其扫描,检测范围为400cm-1~4000cm-1。
2 结果与讨论
2.1 活性评价结果
表1为不同浓度磷钨酸改性的催化剂反应产物的时空收率。从表中可以看出,经磷钨酸改性后乙醇和乙酸的时空收率均有较大变化,随着磷钨酸浓度的不断增加,乙酸的时空收率呈先增大后减小的趋势;除CAT4催化剂之外,其余催化剂几乎没有乙醇生成,CAT4催化剂总时空收率最高,为10.54mg/(g·h)。评价结果表明用磷钨酸对Co-Pd/TiO2催化体系进行改性的确可以提高催化剂的活性,尤其对乙醇的生成极为突出,这与之前相关研究的结论相一致[15],适宜浓度的磷钨酸有利于CH4的解离吸附和氧化偶联。
图1为不同浓度磷钨酸改性的催化剂反应产物选择性分布图。从图中可以看出,各催化剂产物选择性波动较大,尤其是乙醇的选择性变化较大,在磷钨酸浓度为0.04mol/L时乙醇的选择性最高,达到39.7%,这表明磷钨酸对乙醇的生成有促进作用。

表1 磷钨酸改性催化剂的产物时空收率Table 1 Formation rates of products over different catalysts
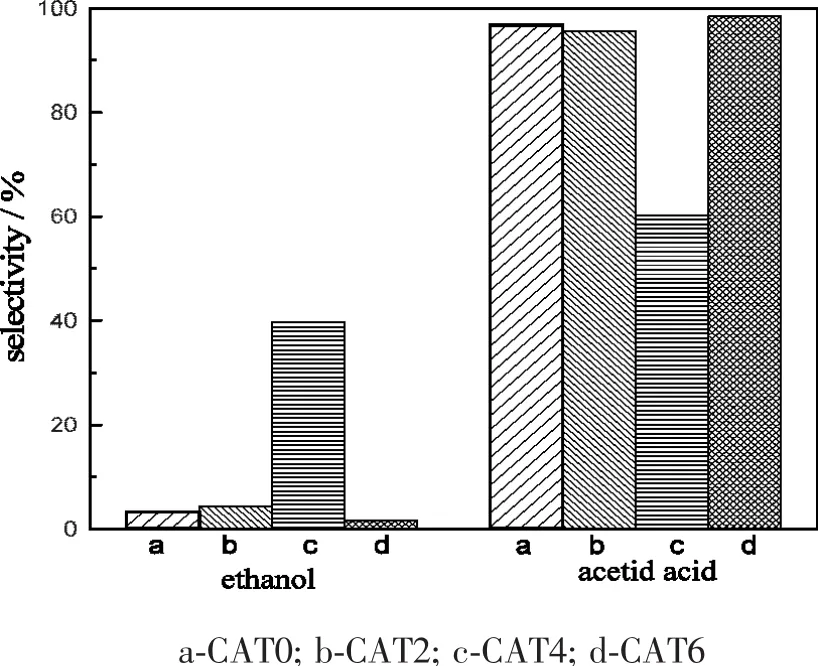
图1 磷钨酸改性催化剂反应产物选择性分布图Fig.1 Distribution of products over different catalysts
2.2 催化剂XRD表征

图2 磷钨酸改性催化剂反应前后的XRD谱图Fig.2 XRD patterns of different catalysts before and after reaction
图2为不同浓度磷钨酸改性的催化剂反应前后的XRD谱图。从反应前的XRD谱图中可以看出, 所有催化剂均在 25.3°、37.5°、47.6°、55.1°和63.1°出现了衍射峰,可归属于锐钛矿型特征衍射峰。通过对比发现,随着磷钨酸浓度的不断增加锐钛矿型衍射峰先弥散后尖锐,说明磷钨酸改性会造成锐钛矿的结构发生一定幅度的转变。所有催化剂中钴物种均以CoTiO3形式存在,未出现钴的氧化物的衍射峰,这可能是由于钴的氧化物与载体TiO2发生强相互作用引起的;钯物种均以PdO形式存在。对比发现,随着磷钨酸浓度的不断增加钴物种和钯物种衍射峰强度均先减弱后增强,CAT4催化剂中钴物种和钯物种衍射峰基本消失,说明磷钨酸改性可以影响活性金属的分散性[17,18]。以上分析表明,催化剂经过适宜浓度的磷钨酸改性,会使活性金属高度分散,这可能是磷钨酸改性可提高催化剂活性的原因。
从反应后的XRD谱图中可以看出,与反应前对比,各催化剂中归属为CoTiO3的衍射峰均变尖锐,说明反应后钴物种发生团聚;各催化剂中基本没有出现与钯物种相关的衍射峰,这可能是因为反应后活性金属流失使得Pd含量较低,没有达到仪器可检测的范围。以上分析表明,催化剂失活的原因可能是活性金属发生团聚。
2.3 催化剂NH3-TPD-MS表征
催化剂的表面酸性主要取决于催化剂的载体而非活性金属[19],TiO2本身具有一定的酸性,但其酸性较弱,而磷钨酸属于超强酸,其酸性较强,通过磷钨酸改性后的催化剂表面的酸性一定会发生改变。NH3-TPD-MS表征主要是用来测定催化剂表面的酸性。催化剂表面的酸性中心强弱与升温脱附时的温度相关联,根据文献[20]报道,一般将催化剂表面的酸性中心分为:弱酸位(≤200℃)、中强酸位(200~400℃)和强酸位(≥400℃)。
图3为不同浓度磷钨酸改性的催化剂反应前的NH3-TPD-MS谱图。从图中可以看出,CAT0催化剂分别在 162℃、285℃、367℃和 649℃附近出现了四个脱附峰,分别对应的是一个弱酸中心、两个中强酸中心和一个强酸中心,并且在162℃附近出现的脱附峰峰面积最大,说明CAT0催化剂表面弱酸酸量最大;CAT2催化剂分别在 162℃、339℃、432℃、517℃和621℃附近出现了五个脱附峰,分别对应的是一个弱酸中心、一个中强酸中心和三个强酸中心,催化剂表面强酸酸量明显增加;CAT4催化剂分别在 162℃、264℃、347℃和 509℃附近出现了四个脱附峰,分别对应的是一个弱酸中心、两个中强酸中心和一个强酸中心;CAT6催化剂只有在544℃和660℃附近出现了两个脱附峰,对应的是两个强酸中心,但没有弱酸中心和中强酸中心出现。
对比四个催化剂的NH3-TPD-MS谱图可以发现,随着磷钨酸浸渍浓度的不断增加,催化剂表面弱酸中心酸量逐渐减少,强酸中心酸量逐渐增加,而表面中强酸中心酸量除CAT6催化剂之外其余三个催化剂没有发生太大变化,说明适宜浓度的磷钨酸改性并不会对催化剂表面中强酸量产生太大影响,只是会改变催化剂表面的弱酸量和强酸量,且随着磷钨酸浓度的改变呈规律性变化,但是当磷钨酸浓度过大时则不利于表面中强酸位的形成。
结合活性评价数据和上述表征分析结果可以得出:催化剂表面中强酸量不是影响催化活性的唯一因素。

图3 磷钨酸改性催化剂反应前的NH3-TPD-MS谱图Fig.3 NH3-TPD-MS profiles of different catalysts beforereaction
2.4 催化剂N2-吸附表征
图4 为不同浓度磷钨酸改性的催化剂反应前后N2吸附-脱附等温线及BJH孔径分布图。从图4可以看出,所有催化剂均表现为IV型吸附-脱附等温线,说明这四个催化剂均为介孔材料。由孔径分布图可以看出,所有催化剂孔径分布主要集中在2.5~8nm 之间。

图4 磷钨酸改性催化剂反应前后N2吸附-脱附等温线及BJH孔径分布图Fig.4 N2 adsorption-desorption isotherms and pore size distributions of catalysts before and after reaction
表2为各催化剂反应前后织构参数。从表2可以看出,随着改性催化剂磷钨酸浓度的不断增加催化剂的比表面积先增大后减小,这与催化剂的活性评价结果相一致。
比较CAT2和CAT4两个催化剂,各织构参数变化不大。结合活性评价结果,两个催化剂的乙酸生成量几乎相同,但CAT4催化剂有较多的乙醇生成。虽然CAT2催化剂有适宜的孔道结构,但乙醇的选择性较低,说明该系列催化剂产物中的乙醇可能不是通过乙酸加氢反应合成的。那么,该系列催化剂产物中的乙醇最有可能是由于CH4氧化偶联途径形成的[21]。这与文献中的研究结果相一致。
比较CAT0和CAT4催化剂,最可几孔径基本保持不变,CAT4催化剂的孔容和比表面积最大。结合活性评价数据,CAT4催化剂的乙酸生成量最大,可能是因为其较大的比表面积为乙酸的生成提供了更多的活性位点,从而促进了乙酸的生成,说明催化剂的比表面积是影响乙酸产率的关键因素。
综合上述分析,同时增大催化剂的比表面积和表面中强酸量是提高乙酸产率的因素。

表2 磷钨酸改性催化剂反应前(B)后(A)的织构参数Table 2 Textural properties of different catalysts before(B)and after(A)reaction
2.5 催化剂FT-IR表征
图5为CAT2和CAT4催化剂反应前的红外光谱图。从图中可以看出,在3441cm-1和1633cm-1附近存在着吸收峰,二者均对应O-H键的振动,这可能是由于催化剂的表面吸附了一定量的水所致[22,23]。在2969cm-1处存在C-H键的吸收峰,可能是由于有机物未全部分解所致。在507cm-1附近存在的吸收峰为Ti-O-Ti的振动吸收峰[24]。从图中还可以看出,CAT4催化剂的TiO2吸收峰发生了明显偏移,这可能是由于Ti-O-Ti键与磷钨酸产生相互作用使其弯曲振动受到影响,由此可以推测催化剂中的TiO2与磷钨酸之间有化学键生成,这与文献[15]报道相一致。

图5 磷钨酸改性催化剂反应前的FTIR谱图Fig.5 FT-IR spectra of catalysts modified by different concentration phosphotungstic acid
2.6 催化剂XPS表征
图6 为CAT2和CAT4催化剂反应前后Co2p的XPS谱图。表3为CAT2和CAT4催化剂反应前后的表面各元素结合能。结合图6和表3可以看出,两个主峰均在高结合能端有明显的伴峰出现,归属于Co2+的2p3/2和2p1/2[25]。CAT2催化剂在反应前后 Co2p3/2的结合能分别在 781.3eV和 781.9eV,CAT4催化剂在反应前后Co2p3/2的结合能分别在781.8eV和782.3eV,对应的Co 2p3/2的结合能均高于 CoO 中 Co2p3/2的结合能(780.1eV±0.3eV)[26-28],这可能是因为在催化剂制备过程中活性金属与TiO2载体之间存在强烈的相互作用所致,这与XRD表征结果相一致,它们之间形成了CoTiO3物种。同时两个主峰宽化程度较明显,说明Co所处电子环境较复杂[29],需要进一步分析研究来确定。
由表3可知,反应前的两个催化剂的Pd 3d5/2结合能分别在337.1eV和337.7eV,可归属于PdO[30];反应后对应的Pd3d5/2的结合能都在335.8eV附近,可归属于金属单质Pd[29],这表明在反应过程中催化剂中的Pd物种主要是以Pd0的形式存在。反应后的两个催化剂Pd3d5/2的结合能分别在336.2eV和336.6eV,均高于 Pd0 的标准结合能(335.8eV)[30],均为给电子环境,说明金属单质Pd所处的电子环境与杂多酸浓度有一定关系。同样,反应后的两个催化剂Co2p3/2结合能较之反应前均有所增大,说明杂多酸对Co2+的存在形态及所处环境均有较大影响。结合活性评价数据和上述分析可知,活性物种处于吸电子环境可能有利于催化剂活性的提高。
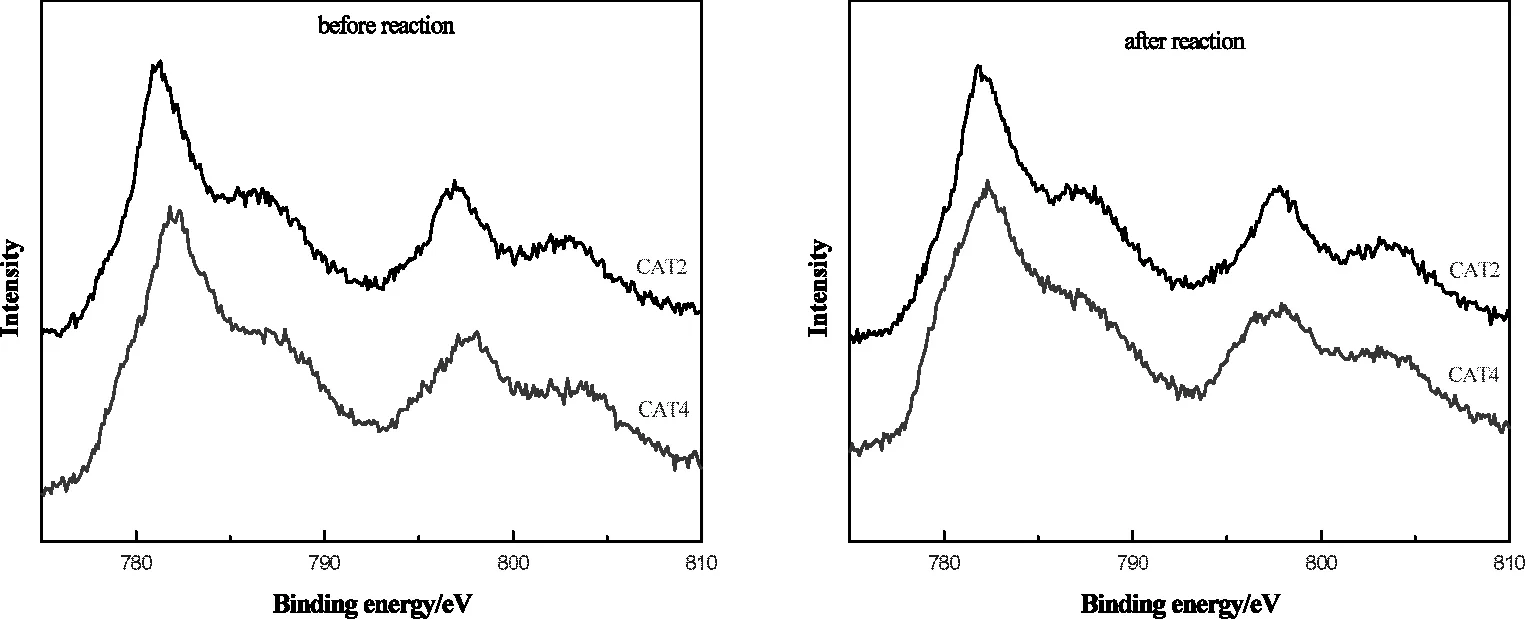
图6 磷钨酸改性催化剂反应前后Co2p的XPS谱图Fig.6 Co2p XPSspectra of catalysts CAT2 and CAT4 before and after reaction
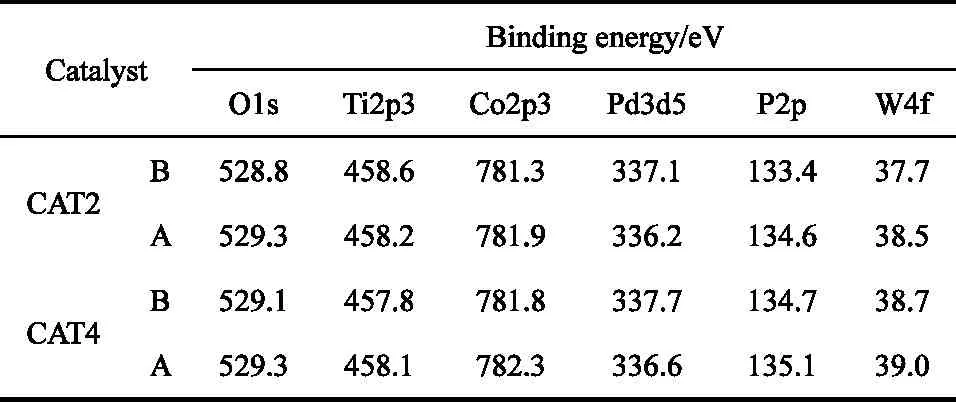
表3 磷钨酸改性催化剂反应前(B)后(A)表面各元素的结合能Table 3 Binding energy of every element on the surfaces of catalyst CAT2 and CAT4 before(B)and after(A)reaction
表4为CAT2和CAT4催化剂反应前后表面各元素原子含量。由表可知:CAT4催化剂反应前表面C含量较低,说明一定浓度的磷钨酸改性对催化剂的表面性质有较大影响。另外CAT4催化剂表面Co含量较多,Co/Pd比更大,催化剂活性较好,说明相对多的Co有利于CH4-CO2的两步梯阶反应。而Co有利于CH4的活化表明CH4活化在该反应工艺过程中处于主导地位[31],需要更多的活性中心来活化甲烷。但是,两个催化剂的Co/Pd物质的量比远大于初始投料组成[n(Co)/n(Pd)=3.61]。分析推测Co/Pd比变大的原因:(1)催化剂活性组分流失;(2)催化剂活性组分分布不均匀。该推测需要进一步对催化剂研究验证。
3 结论
以上研究表明,杂多酸改性的Co-Pd/TiO2催化剂在CH4-CO2两步梯阶转化过程中不仅可以得到高选择性的乙酸,而且适宜浓度杂多酸改性的催化剂还有利于乙醇的生成。在常压、150℃的反应条件下,以0.04mol/L磷钨酸改性的催化剂乙醇和乙酸的总时空速率达到10.54mg/(g·h),与未经磷钨酸改性的催化剂对比活性提高了2.55倍。这是因为:
(1)适宜浓度的磷钨酸改性有利于提高催化剂中活性金属的分散性,高分散的Co和Pd有利于催化活性的提高;
(2)催化剂比表面积和表面中强酸量的协同作用是影响催化活性的关键因素,适宜浓度的磷钨酸改性有利于催化剂表面中强酸位的形成和酸量的增加;
(3)磷钨酸改性催化剂产物中的乙醇主要是通过CH4氧化偶联反应途径合成的。
(4)适宜浓度的磷钨酸改性使催化剂中的TiO2与磷钨酸间生成了某种化学键,促进了CH4的活化并提高了催化活性;
(5)活性物种处于吸电子环境可能有利于催化剂活性的提高。
猜你喜欢
中学生数理化·高一版(2022年4期)2022-05-09 15:36:00
井冈山大学学报(自然科学版)(2022年2期)2022-03-31 02:39:50
环境科技(2016年1期)2016-11-08 12:17:32
浙江大学学报(工学版)(2016年9期)2016-06-05 09:20:57
当代化工研究(2016年5期)2016-03-20 16:21:29
化学研究(2015年3期)2015-11-27 05:39:50
橡胶工业(2015年8期)2015-02-23 23:41:15
石油化工(2015年11期)2015-02-05 08:26:01
理科考试研究·高中(2014年11期)2014-11-26 22:15:18
江西理工大学学报(2013年1期)2013-03-20 14:57:07
