
长期施肥和灌溉对土壤细菌数量、多样性和群落结构的影响
2018-02-04 03:06杨亚东王志敏曾昭海
中国农业科学 2018年2期
杨亚东,王志敏,曾昭海
长期施肥和灌溉对土壤细菌数量、多样性和群落结构的影响
杨亚东,王志敏,曾昭海
(中国农业大学农学院, 北京 100193)
【目的】通过对华北地区一年两熟种植模式下冬小麦生长季不同施肥和灌溉处理下土壤细菌群落的研究,揭示长期不同施肥和灌溉制度下土壤细菌数量、多样性和群落结构的变化规律。为科学施肥和灌溉,提高农田地力和维持土壤微生物多样性等提供依据。【方法】依托中国农业大学吴桥实验站,选取长期施肥和灌溉定位试验的6个处理冬小麦收获后耕层土壤为研究对象,分别为化肥+不灌溉(CI0)、化肥+拔节期灌溉(CI1)、化肥+拔节期灌溉+灌浆期灌溉(CI2)、有机肥+不灌溉(MI0)、有机肥+拔节期灌溉(MI1)和有机肥+拔节期灌溉+灌浆期灌溉(MI2)。借助荧光定量PCR技术和Illumina Miseq高通量测序平台,以16S rRNA基因为标靶,研究长期不同施肥和灌溉制度对土壤细菌数量、多样性和群落结构的影响,并分析细菌数量、多样性和群落结构变化与土壤理化性质的相关性。【结果】灌溉显著提高了土壤含水量和土壤pH,施有机肥比施化肥显著提高了土壤有机碳含量。不同处理细菌16S rRNA基因拷贝数为每克干土4.34×109—1.39×1010。灌溉显著提高了细菌数量,化肥和有机肥处理分别提高了1.17—1.60和0.76—1.93倍。多样性指数结果表明灌溉显著影响细菌群落多样性指数,施肥对细菌群落多样性指数的影响均不显著。门水平上,18个样品共获得39个类群,其中变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、绿弯菌门(Chloroflexi)、酸杆菌门(Acidobacteria)和拟杆菌门(Bacteroidetes)为优势类群,相对丰度共占77.22%—86.28%。不同处理间放线菌门(11.09%—27.01%)、拟杆菌门(5.45%—12.13%)和Saccharibacteria(2.41%—3.77%)的相对丰度差异显著。灌溉显著降低了放线菌门和Saccharibacteria的相对丰度,化肥和有机肥处理分别降低了36.48%—48.03%、22.17%—33.67%和15.21%—45.54%、13.40%—23.97%。层次聚类和主成分分析结果显示施肥和灌溉对细菌群落结构都产生影响,相同灌溉次数处理的细菌群落结构相似,而相同施肥处理间细菌群落结构差异较大,表明灌溉对细菌群落结构的影响强于施肥。此外,土壤含水量、土壤pH、全氮含量和有机碳含量与细菌数量、多样性指数和群落结构存在一定的显著相关关系。【结论】灌溉显著改变了细菌数量、多样性和群落结构,施肥对细菌数量和群落结构的影响较小。土壤含水量和土壤pH是造成土壤细菌数量、多样性和群落结构差异的主要原因。
有机肥;灌溉;细菌群落多样性;高通量测序;华北地区
0 引言
【研究意义】土壤微生物是土壤肥力的重要组成部分,在维持土壤生态功能中扮演着重要角色[1],同时也是评价土壤质量和生产力的重要指标[2]。施肥和灌溉是农业生产中最主要的农业管理措施,具有促进作物生长和提高产量的作用,同时也对土壤微生物产生影响。土壤微生物数量、群落结构和生物活性的变化直接或间接影响土壤肥力和可持续生产力[3]。华北地区是中国重要的粮食生产基地,以冬小麦-夏玉米种植模式为主。冬小麦生长季需要大量施肥和抽取地下水灌溉,导致地下水污染和地下水水位持续下降[4],使该地区面临严峻的水资源危机。研究不同施肥和灌溉处理土壤细菌数量、多样性和群落结构的变化,揭示土壤微生物对施肥和灌溉的响应机制,为改进施肥和灌溉制度,提高土壤肥力和生产力,缓解地下水资源危机提供依据。因此,开展施肥结合灌溉对土壤细菌数量、多样性和群落结构影响的研究具有重要意义。【前人研究进展】施肥改变了土壤微生物的数量、群落结构和活性,引起土壤生物肥力差异,进而影响土壤结构、肥力和可持续生产力[5]。长期施用化肥显著提高了水稻土壤中细菌数量[6],并改变了细菌群落结构和多样性[6-7]。施用有机肥能显著改变微生物的群落结构及多样性[8-9],提高微生物的生物量和代谢活性[10]。此外,土壤水分管理也对土壤微生物的数量、群落结构和活性产生影响[11]。土壤含水量通过影响土壤营养物质运输、底物可用性和土壤其他特性,改变土壤微生物的群落组成和活性[12-13]。近年来,随着测序技术的快速发展,新一代测序技术被广泛应用到环境微生物群落的研究中。高通量测序技术具有测序深度深和获得数据量大等优点,能更真实地揭示微生物群落的复杂性和多样性,加快了环境中不可培养和痕量微生物的研究[14]。前人利用Roche 454和Illumina Miseq测序平台探讨了施肥和灌溉等农业管理措施对不同类型土壤中微生物群落多样性和结构组成的影响[8, 15-17],并取得了一定进展。【本研究切入点】由于冬小麦生长季长期施用化肥和抽取地下水灌溉,造成了华北地区土壤氮素淋溶、生产力降低和严峻的地下水资源危机[4,18]。施用有机肥能改善土壤质量、改变土壤微生物组成,提高土壤生产力[19]。同时,减少冬小麦生长季灌溉用水是缓解华北地区地下水超采压力的重要措施。先前的研究多集中在施肥或灌溉等单一措施对土壤细菌群落的研究,缺乏施肥与灌溉结合管理对土壤细菌群落影响的系统研究。【拟解决的关键问题】本研究以中国农业大学吴桥实验站长达11年的冬小麦-夏玉米种植模式定位试验为平台,以冬小麦收获后耕层土壤为研究对象,以16S rRNA基因为标靶,借助荧光定量PCR技术和Illumina MiSeq高通量测序平台,探讨不同肥料类型和灌溉次数处理土壤细菌数量、多样性和群落组成的变化及其与土壤理化性质的关系,为优化该地区农田施肥和灌溉制度,提高土壤肥力,维护土壤微生物生态系统和农业可持续发展等方面提供依据。
1 材料与方法
1.1 试验设计
试验在中国农业大学吴桥实验站范屯乡姚庄村(北纬37°41',东经106°37')进行。试验区位于沧州地区最南端,海河平原黑龙港流域中部,属暖温带半湿润大陆性季风气候,全年光照2 724.8 h,年平均气温12.9 ℃,无霜期201 d,历年平均年降水量562 mm,小麦季平均降雨量127.3 mm。土壤为冲积型盐化潮土,壤质底黏。
施肥和灌溉长期定位试验起始于2005年,设2种肥料类型和3个灌溉梯度,共6个处理。分别为:化肥+不灌溉(CI0)、化肥+拔节期灌溉(CI1)、化肥+拔节期灌溉+灌浆期灌溉(CI2)、有机肥+不灌溉(MI0)、有机肥+拔节期灌溉(MI1)和有机肥+拔节期灌溉+灌浆期灌溉(MI2)。小区面积为66.7 m2,3次重复。冬小麦播种前浇足底墒水,灌溉量为750 m3·hm-2,之后每次灌溉量均为750 m3·hm-2。冬小麦生长季氮、磷、钾肥施用量分别为尿素150 kg·hm-2、磷酸二铵300 kg·hm-2和硫酸钾225 kg·hm-2;有机肥为鸡粪,施用量为22.5 m3·hm-2。所有肥料均作为基肥一次性施入。田间管理同常规大田生产。试验开始前耕层(0—20 cm)土壤的基本理化性质:pH 7.43,有机碳11.10 g·kg-1,全氮1.05 g·kg-1,有效磷49.10 mg·kg-1,速效钾123.75 mg·kg-1。
1.2 土壤样品采集
于2016年6月,取冬小麦收获后耕层(0—20 cm)土壤。每个小区随机取5个样点混合成1个样品,用冰盒保存土样,运输至实验室。将土壤样品过2 mm筛,去除小麦根系残体和杂物后分为两部分。一部分冻存于-80℃冰箱中,用于土壤微生物分析。另一部分鲜土样用于测定土壤铵态氮、硝态氮和含水量,余下部分自然风干后测定土壤pH、全氮和有机碳。
1.3 试验方法
1.3.1 土壤理化性质测定 土壤含水量采用烘干法测定,土壤pH采用电位法测定(水土比=2.5﹕1),土壤全氮含量采用半微量凯氏定氮法测定,土壤总有机碳测定采用浓硫酸-重铬酸钾消煮-硫酸亚铁滴定法[20]。土壤铵态氮和硝态氮用2 mol·L-1KCl溶液浸提鲜土样后,采用流动分析仪测定(SAN++,Skalar,Holland)。
1.3.2 土壤微生物总DNA提取和PCR扩增 土壤总DNA使用美国OMEGA公司的E.Z.N.A.®Soil DNA Kit(Omega, GA, USA)试剂盒,每个样品称取约0.5 g新鲜土壤,按照试剂盒提取步骤进行。用1%的琼脂糖凝胶电泳检测提取DNA的纯度和完整性,用核酸定量仪(NanoDrop ND-1000)检测提取DNA的浓度和纯度。
用引物BACT1369F(5′-CGG TGA ATA CGT TCY CGG-3′)和PROK1541R(5′-AAG GAG GTG ATC CRG CCG CA-3′)[21]扩增细菌16S rRNA基因。PCR体系为:10×缓冲液 5.0 μL,dNPT(2.5 μmol·L-1)4.0 μL,上下游引物(10 μmol·L-1)各1.0 μL,DNA模板2.0 μL(1-10 ng),(5 U·μL-1)1.0 μL(TaKaRa,大连,中国),最后用超纯水(ddH2O)补至50.0 μL。PCR扩增条件为:95℃ 3 min;94℃ 45 s,55℃ 30 s,72℃ 45 s,30个循环;72℃ 5 min。将PCR产物回收,连接至pMD18-T载体(TaKaRa,大连,中国),转化至大肠杆菌DH5α感受态中,筛选阳性克隆测序,并提取含16S rRNA基因的质粒,用核酸定量仪(NanoDrop ND-1000)测定质粒浓度,计算16S rRNA基因拷贝数,按10倍梯度稀释至每微升8.75×108—8.75×103拷贝数,共6个梯度,用于制备16S rRNA基因标准曲线。
1.3.3 荧光定量PCR 采用SYBR Green定量PCR法测定细菌16S rRNA基因丰度,反应在ABI 7500荧光定量PCR仪(ABI,CA,USA)上进行。反应体系为:SYBR®Premix Ex Taq Ⅱ(Thi RNaseH Plus)12.5 μL,上下游引物(10 μmol·L-1)各0.25 μL,ROX Reference Dye(50×)0.5 μL,DNA模板1.0 μL(1—10 ng),最后用超纯水(ddH2O)补至25.0 μL。荧光定量PCR采用两步法进行,条件为:95 ℃ 30 s;95 ℃ 5 s;60 ℃ 1 min,40个循环。荧光定量PCR扩增效率为94.4%,2为0.999。
1.3.4 高通量测序分析 用引物338F(5′-ACT CCT ACG GGA GGC AGC A-3′)[22]和806R(5′-GGA CTA CHV GGG TWT CTA AT-3′)[23]扩增细菌16S rRNA基因V3-V4区,扩增体系和程序同1.3.2所示。每个样品的上游引物5′端添加一段长度为8 bp的特异性多肽(barcode),用于区分样品。回收PCR产物并测定浓度,将同一处理的多个样品混匀,使各处理用于测序的样品DNA浓度一致,采用Illumina MiSeq测序平台进行双末端(Paired-end)测序。测序服务委托上海美吉生物医药科技有限公司完成。
1.4 测序数据处理
对下机原始数据进行质量控制,用软件QIIME 1.8.0对原始序列进行过滤、拼接、去除嵌合体[24],并对序列长度进行筛选。根据特异性多肽将序列确定为最终的样品序列。按照以下要求进行。(1)过滤read尾部质量值20以下的碱基,设置50 bp的窗口,如果窗口内的平均质量值低于20,从窗口开始截去后端碱基,过滤质控后50 bp以下的read;(2)根据PE reads之间的重叠(overlap)关系,将成对reads拼接(merge)成一条序列,最小重叠长度为10 bp;(3)拼接序列的重叠区允许的最大错配比率为0.2,筛选不符合序列;(4)根据序列首尾两端的特异性多肽和引物区分样品,并调整序列方向,特异性多肽允许的错配数为0,最大引物错配数为2,最后获得用于后续分析的高质量序列。
采用UPARSE 7.1软件将优质序列聚类成操作分类单元(Operational Taxonomic Unit,OTU),阈值设置为97%[25]。采用RDP-classifier贝叶斯算法[26]对97%相似水平的OTU代表序列进行注释,得到每个OTU的分类学信息。将不能聚类在任何分类水平已知类群的序列定义为未分类。利用Mothur 1.30.1软件在97% 相似水平上进行α多样性指数分析[27]。
1.5 数据分析
细菌16S rRNA基因拷贝数取对数后,用Sigma Plot 12.5软件制图。土壤理化性质、细菌16S rRNA基因拷贝数和α多样性指数等数据的多重比较、方差分析和相关性分析用SPSS 20.0软件完成。细菌群落结构的主成分分析和冗余分析用R 3.3.2软件完成。
2 结果
2.1 不同处理土壤理化性质
长期不同施肥和灌溉处理显著影响土壤理化性质(表1)。方差分析结果表明,施肥显著改变了不同处理间土壤所有理化性质,灌溉显著改变了不同处理间除C/N比(=1.06,0.377)外的其他理化性质,施肥和灌溉的交互作用显著改变了不同处理间除有机碳(=0.467,0.638)外的其他理化性质(表1)。有机肥处理中,灌溉显著提高了土壤含水量(<0.001)和全氮含量(<0.01),分别提高了2.2—6.6个百分点和6.0%—11.0%;化肥处理中,灌溉显著提高了土壤含水量(<0.001)、土壤pH(<0.001)和铵态氮含量(<0.001),分别提高了1.1—4.2个百分点、0.37—0.41个单位和2.7—5.1倍。灌溉次数相同时,施有机肥显著提高了土壤全氮(<0.001)和有机碳含量(<0.001),分别提高了17.8%—27.8%和7.8%—11.7%。
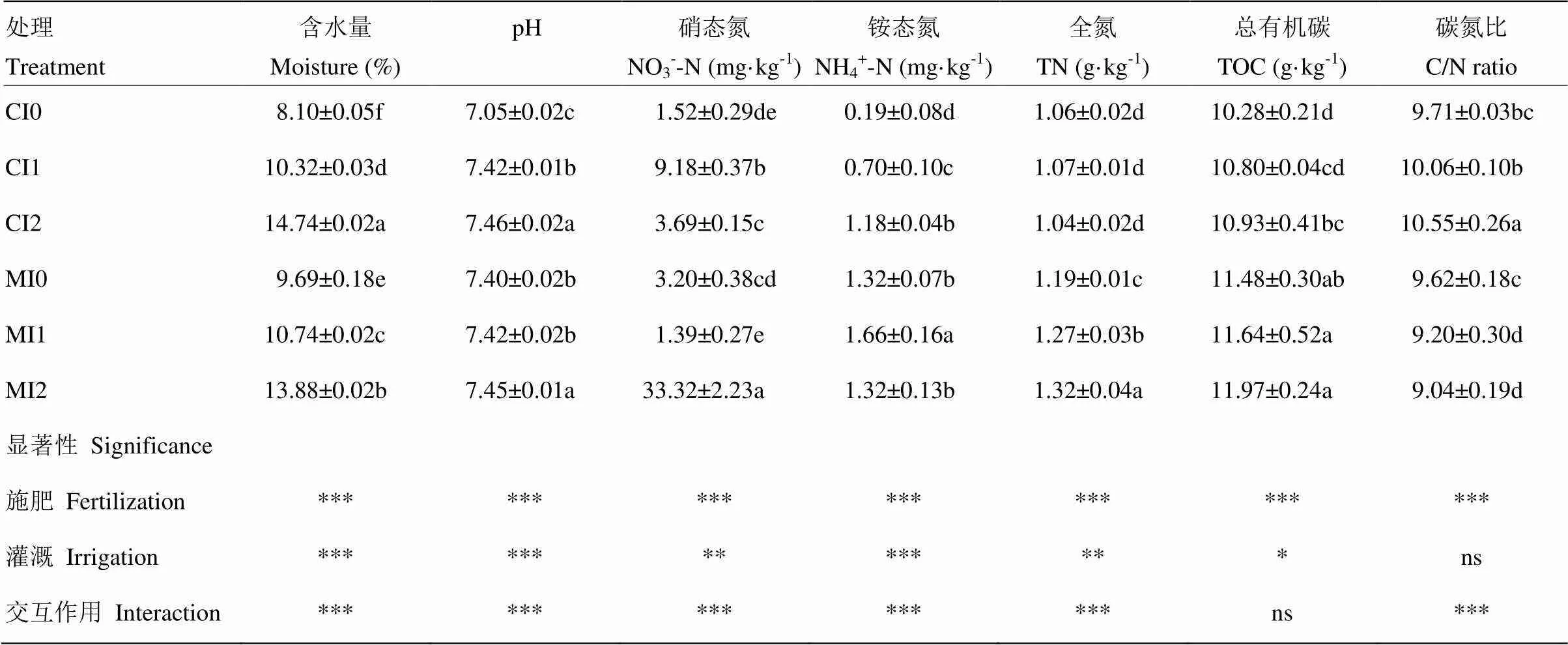
表1 不同施肥和灌溉处理土壤理化性质
数据为平均值±标准差(=3);同列数值后不同小写字母表示差异显著(<0.05)。CI0,化肥+不灌溉;CI1,化肥+拔节期灌溉;CI2,化肥+拔节期灌溉+灌浆期灌溉;MI0,有机肥+不灌溉;MI1,有机肥+拔节期灌溉;MI2,有机肥+拔节期灌溉+灌浆期灌溉。*表示<0.05,**表示<0.01,***表示<0.001,ns表示差异不显著。下同
Data are means±standard deviation (n=3); Different small letters mean significant differences (<0.05). CI0, chemical fertilizer + no irrigation; CI1, chemical fertilizer + irrigated at the jointing stage; CI2, chemical fertilizer + irrigated at the jointing and filling stages; MI0, manure fertilizer + no irrigation; MI1, manure fertilizer + irrigated at the jointing stage; MI2, manure fertilizer + irrigated at the jointing and filling stages. *<0.05, **<0.01, ***<0.001, ns: not significant difference. The same as below
2.2 不同处理细菌16S rRNA基因丰度
不同施肥和灌溉处理中土壤细菌16S rRNA基因拷贝数为每克干土4.34×109—1.39×1010(图1)。方差分析结果表明,灌溉(=137.74,<0.01)及施肥与灌溉的交互作用(=8.92,<0.05)显著影响细菌16S rRNA基因拷贝数(表2)。灌溉显著增加了细菌16S rRNA基因拷贝数(<0.05),其中处理MI2和MI1比MI0分别提高了192.6%和75.9%,处理CI2和CI1比CI0分别提高了160.0%和126.8%。灌溉相同时,处理MI2中细菌16S rRNA基因拷贝数显著高于CI2(<0.05),处理MI1和CI1、MI0和CI0间差异则不显著(>0.05)。
相关性分析结果显示,细菌16S rRNA基因拷贝数与土壤含水量(=0.856,<0.001)、土壤pH(=0.675,<0.01)和全氮含量(=0.723,<0.01)均呈显著正相关关系(表3)。
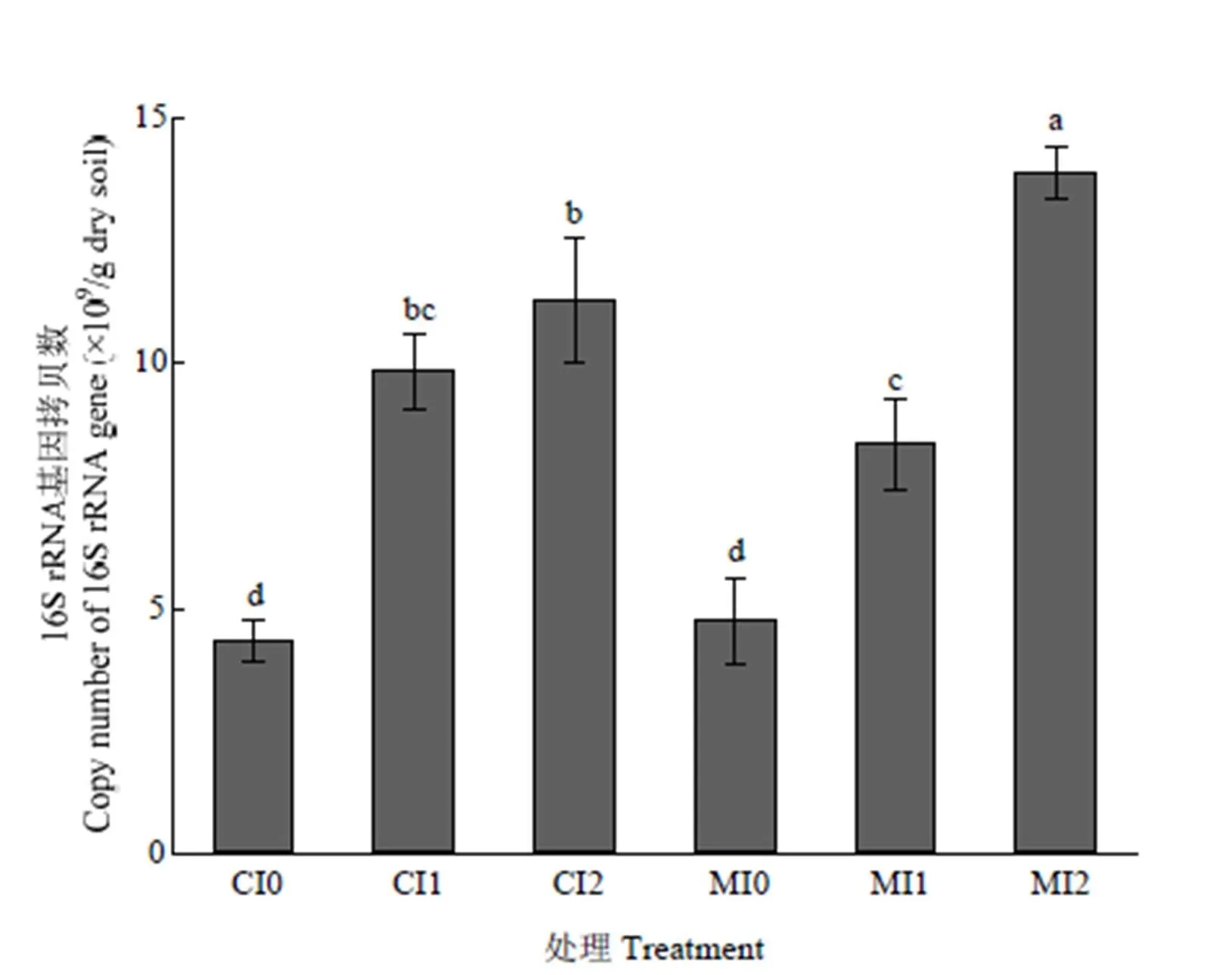
数据为平均值±标准差(n=3);不同小写字母表示差异显著(P<0.05)
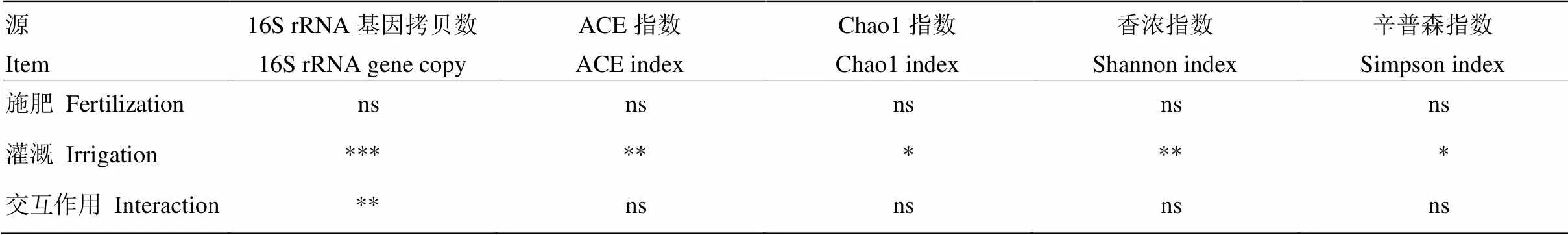
表2 施肥、灌溉及施肥与灌溉的交互作用对细菌16S rRNA基因拷贝数和群落α多样性指数的影响
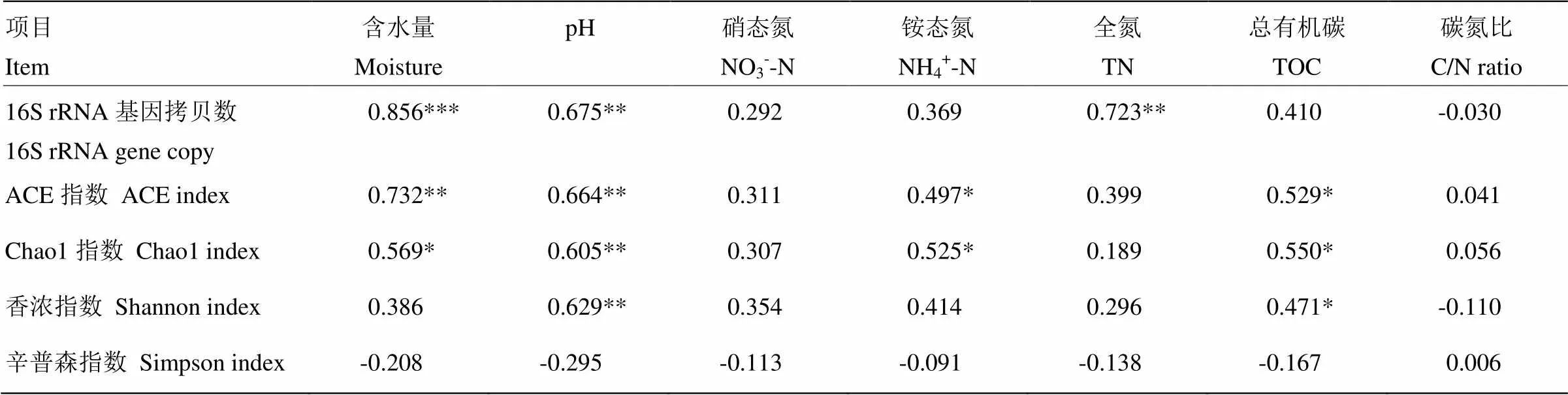
表3 细菌16S rRNA基因丰度、群落α多样性指数与土壤理化性质的Pearson相关性分析
2.3 测序结果和多样性指数
利用Illumina MiSeq平台对细菌16S rRNA基因V3-V4区测序,共得到有效序列707 759条,平均长度为438.16 bp。经抽平筛选后,每个样品得到21 230条高质量序列,包含2 336—2 671个操作分类单元(OTU),各样品的测序覆盖度为0.9657—0.9698(表4)。样品OTU稀释性曲线均趋于平坦,表明测序深度包括了样品中的绝大多数细菌类型,测序数据量合理。
不同处理的ACE指数为2 999—3 178,Chao1指数为3 016—3 168,香浓指数为6.84—6.98,辛普森指数为0.0019—0.0025(表4)。方差分析结果表明,灌溉显著影响ACE指数(=10.484,<0.05)、Chao1指数(=4.216,<0.05)、香浓指数(=14.298,<0.01)和辛普森指数(=5.137,<0.05)(表2)。与不灌溉相比,灌溉处理中ACE指数、Chao1指数和香浓指数分别提高了3.0%—5.9%、2.3%—5.1%和1.2%—2.0%,辛普森指数降低了7.1%—22.7%(表4)。
相关性分析结果显示,ACE指数与土壤含水量(=0.732,<0.01)、土壤pH(=0.664,<0.01)、铵态氮含量(=0.497,<0.05)和有机碳含量(=0.529,<0.05)均呈显著正相关关系。Chao1指数与土壤含水量(=0.569,<0.05)、土壤pH(=0.605,<0.01)、铵态氮含量(=0.525,<0.05)和有机碳含量(=0.550,<0.05)均呈显著正相关关系。香浓指数与土壤pH(=0.629,<0.01)和有机碳含量(=0.471,<0.05)均呈显著正相关关系(表3)。
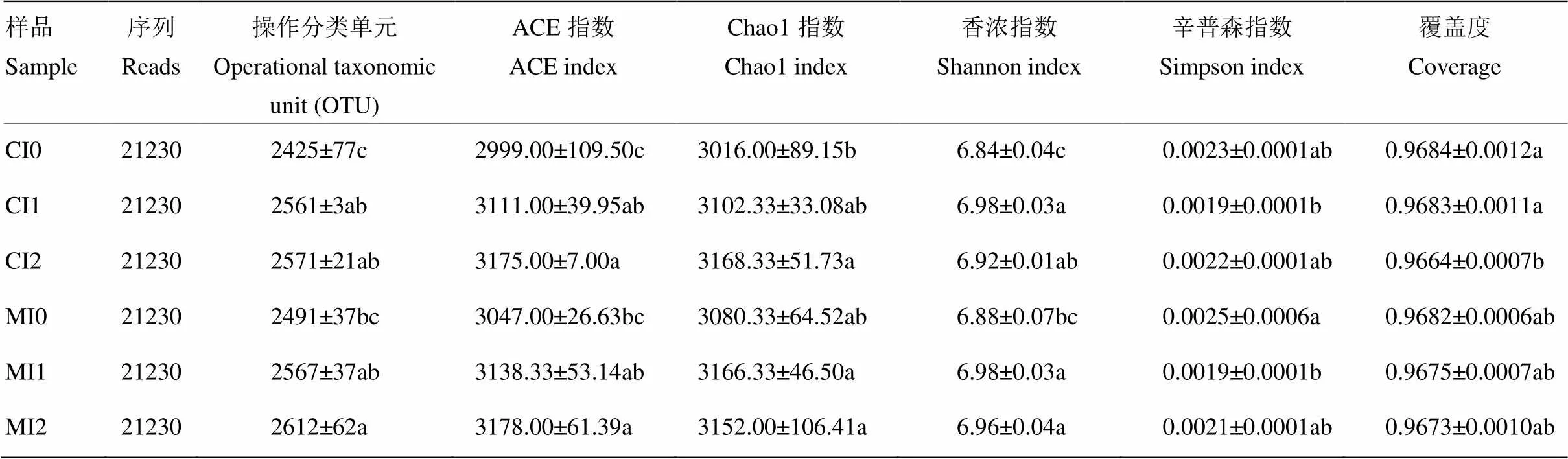
表4 不同施肥和灌溉处理细菌高通量测序结果统计
2.4 不同处理细菌的群落组成
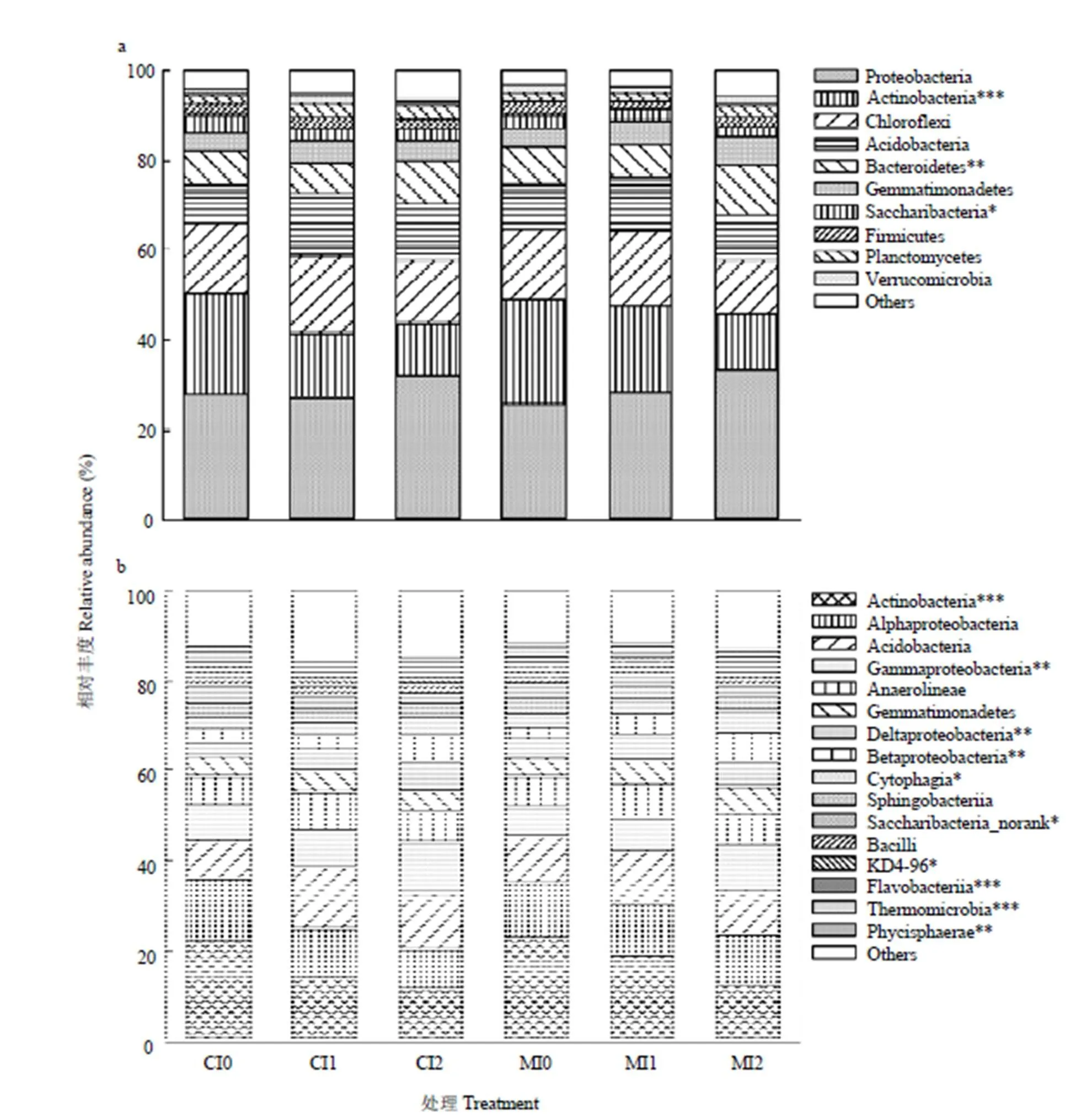
在门水平上,共获得39个类群。将平均相对丰度<1%类群归类为其他,得到17个类群(图2-a)。其中,变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、绿弯菌门(Chloroflexi)、酸杆菌门(Acidobacteria)和拟杆菌门(Bacteroidetes)为主要优势类群,相对丰度分别为22.82%—39.94%、11.09%—27.01%、8.93%—19.86%、5.01%—17.81%和5.45%—12.13%,合计为77.22%—86.28%。不同处理间放线菌门(<0.001)、拟杆菌门(<0.01)和Saccharibacteria(<0.05)的相对丰度差异显著。施肥显著改变了放线菌门(=6.617,<0.05)的相对丰度,灌溉显著改变了放线菌门(=40.946,<0.001)、拟杆菌门(=15.649,<0.001)和Saccharibacteria(=9.772,<0.01)的相对丰度。
在纲水平上,共获得97个类群。将平均相对丰度<1%类群归类为其他,得到17个类群(图2-b)。其中,放线菌纲(Actinobacteria)、-变形菌纲(Alphaproteobacteria)、酸杆菌纲(Acidobacteria)、-变形菌纲(Gammaproteobacteria)和厌氧绳菌纲(Anaerolineae)为主要优势类群,相对丰度分别为11.09%—27.01%、8.83%—17.42%、5.01%—17.81%、6.02%—11.92%和3.24%—9.20%,合计为48.04%—61.65%。不同处理间放线菌纲(<0.001)、-变形菌纲(<0.01)、-变形菌纲(Deltaproteobacteria)(<0.01)、-变形菌纲(Betaproteobacteria)(<0.01)、纤维菌纲(Cytophagia)(<0.05)、Saccharibacteria_norank(<0.05)、KD4-96(<0.05)、黄杆菌纲(Flavobacteriia)(<0.001)、热微菌纲(Thermomicrobia)(<0.001)和Phycisphaerae(<0.001)的相对丰度差异显著。施肥显著改变了放线菌纲(=6.617,<0.05)和黄杆菌纲(F=6.166,<0.05)的相对丰度,灌溉显著改变了放线菌纲(=40.946,<0.001)、-变形菌纲(=15.465,<0.001)、-变形菌纲(=14.427,<0.01)、-变形菌纲(=21.22,<0.001)、纤维菌纲(=10.287,<0.01)、Saccharibacteria_norank(=9.772,<0.01)、KD4-96(=8.36,<0.01)、黄杆菌纲(=88.045,<0.001)、热微菌纲(=30.1,<0.001)和Phycisphaerae(=10.213,<0.01)的相对丰度,施肥与灌溉的交互作用显著改变了可热微菌纲(=7.172,<0.01)的相对丰度。
2.5 不同处理细菌的群落结构及其与环境因子的关系
基于OTU的层次聚类分析结果显示,不同施肥和灌溉处理细菌群落结构差异明显,除MI2_2外,其他处理的3个重复都聚类在一起。其中,不灌溉处理(CI0和MI0)、灌溉一次处理(CI1和MI1)和灌溉两次处理(CI2和MI2)距离较近(图3-a)。主成分分析进一步证实了不同处理细菌群落结构差异明显,第一、二主成分轴对细菌群落结构变异的解释量分别为35.44%和15.33%。其中,灌溉条件相同处理(CI0和MI0,CI1和MI1,CI2和MI2)聚集在一起,而肥料类型相同处理(CI0、CI1和CI2,MI0、MI1和MI2)相距较远(图3-b)。这些结果表明灌溉次数对细菌群落结构的影响强于肥料类型。
标星号表示不同处理中的相对丰度差异显著。*表示P<0.05,**表示P<0.01,***表示P<0.001

图3 不同施肥和灌溉处理细菌群落结构的层次聚类树(a)和主成分分析(b)
为进一步分析不同土壤理化性质对细菌群落结构的影响,对细菌群落结构与土壤理化性质进行冗余分析(图4)。结果显示,土壤含水量(<0.001)、土壤pH(<0.001)、全氮含量(<0.01)、铵态氮含量(<0.001)和有机碳含量(<0.01)均显著影响了细菌的群落结构。
3 讨论
3.1 不同施肥和灌溉处理对细菌数量的影响
本研究利用荧光定量PCR分析了不同施肥和灌溉处理土壤细菌数量,不同处理中细菌16S rRNA基因丰度在109—1010个拷贝数,低于长期施肥水稻土中的细菌数量[6],与长期种植小麦的相似类型旱地土壤中细菌数量级一致[28]。袁红朝等[6]研究表明水稻土有机碳含量高(19.56—22.65 g·kg-1),可为微生物提供充足的碳源,具有较高的微生物数量。本研究中与施化肥相比,施有机肥显著提高了土壤有机碳含量,但细菌数量和有机碳含量却无显著相关关系(=0.41,=0.091)。CHEN等[29]研究表明土壤类型显著影响了水稻土氨氧化微生物的数量和群落结构,因此,土壤类型可能是引起细菌在旱地土壤和水稻土中数量差异的原因。灌溉显著增加了细菌数量(<0.001),与刘方春等[30]对不同干旱处理的研究结果相似。原因可能是灌溉提高了土壤含水量和土壤pH,增加了氮元素的可利用性,为细菌生长提供了充足的氮源,促进了细菌繁殖。细菌数量与土壤含水量(=0.856,<0.001)、土壤pH(=0.675,<0.01)和全氮含量(=0.723,<0.01)均呈显著正相关关系,进一步证实这些土壤理化性质显著影响了细菌的数量。此外,施肥(=1.657,=0.22)对细菌数量的影响不显著,而灌溉(=137.74,<0.01)显著改变了细菌数量,表明灌溉对细菌数量的影响强于施肥。
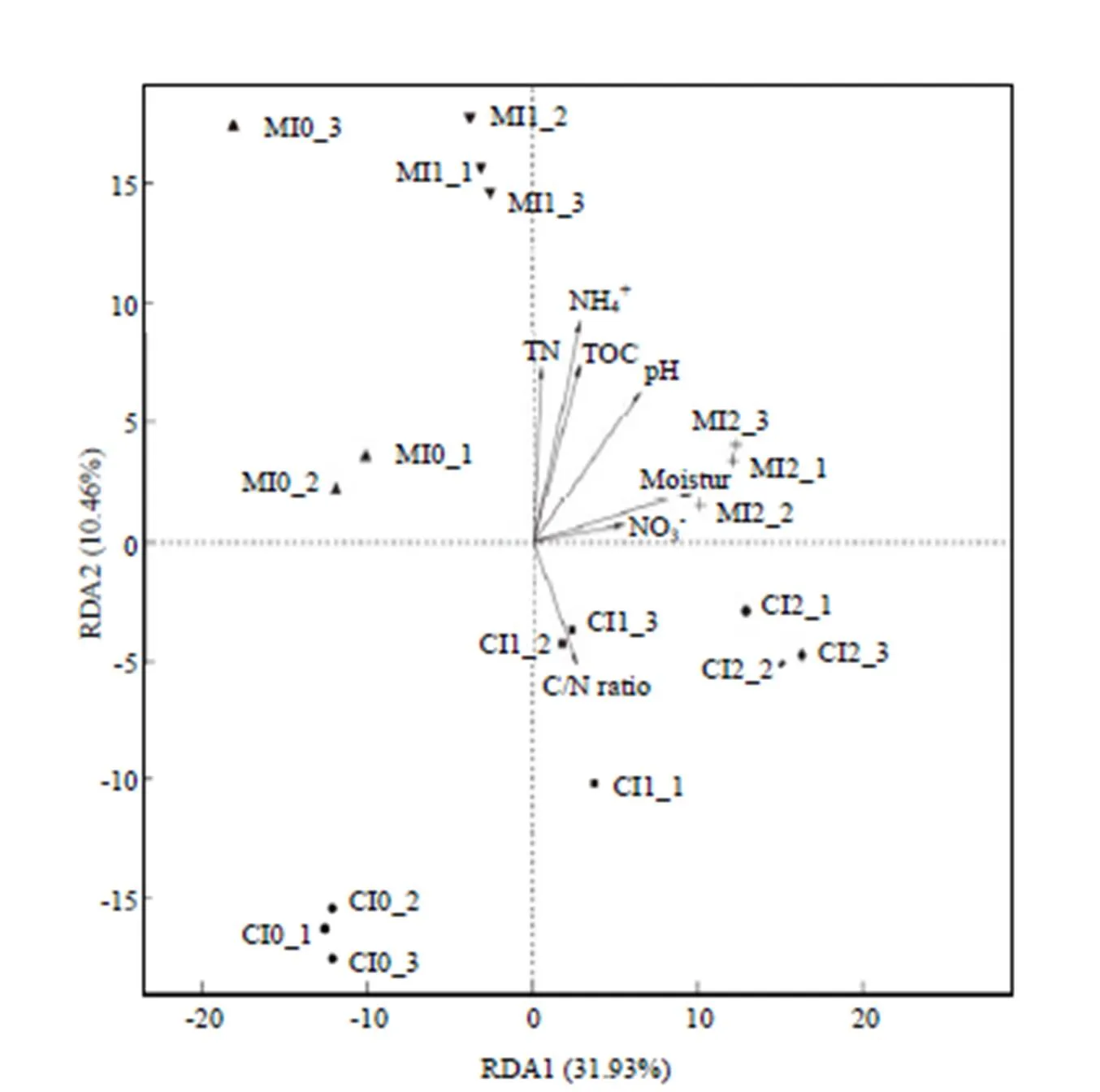
图4 环境因子对细菌群落结构影响的冗余分析
3.2 不同施肥和灌溉处理对细菌多样性的影响
多样性指数是评价细菌群落多样性的重要指标,多样性指数越高表明细菌群落的丰富度和多样性越高。袁红朝等[6]对不同施肥水稻土壤的研究表明施肥和秸秆还田均显著增加细菌多样性,徐永刚等[31]研究表明施有机肥玉米土壤细菌多样性高于施化肥,而本研究中施有机肥和化肥处理细菌群落的ACE、Chao1和香浓指数均差异不显著,可能是由于不同地区土壤性质和作物品种差异造成。刘方春等[30]研究表明适当的干旱可提高樱桃实生苗根际土壤的细菌群落多样性,但过高干旱持续时间过长或者土壤含水量导致细菌群落多样性指数降低,本研究显示灌溉显著增加了细菌群落的ACE、Chao1和香浓指数,表明灌溉增加了细菌的丰富度和多样性,可能是由于灌溉后,土壤含水量增加,在一定的范围内,土壤含氧量合适,氮、磷、钾等无机元素和有机质等可利用性增加,为更多的微生物提供了生长和繁殖的条件。此外,土壤pH是土壤微生物生物量和细菌群落多样性的限制因素[32-33],土壤养分和有机碳含量也是影响细菌多样性的重要因素[33]。本研究中,细菌群落的ACE、Chao1和香浓指数与土壤含水量、pH和有机碳含量存在一定的显著相关关系,表明了土壤含水量、pH和有机碳是影响细菌群落多样性的重要因素。
3.3 不同施肥和灌溉处理对群落组成和结构的影响
细菌是土壤微生物的重要组成,参与了土壤养分循环,而且对维持整个土壤生态系统的稳定性起着重要的作用[34]。本研究利用Illumina Miseq测序平台对6个不同施肥和灌溉处理土壤细菌群落结构进行研究,得到39个门水平、97个纲水平和超过600个属水平上的类群。结果表明,不同处理中优势的门和纲类群相似,但不同处理门和纲水平上的优势类群的相对丰度差异显著(图2-a,2-b)。各处理优势门类群(平均相对丰度>5%)为变形菌门、放线菌门、绿弯菌门和酸杆菌门,与孙瑞波等[15]和LIU等[33]在不同类型农田土壤中得到了细菌优势类群相似,但不同研究中各优势类群的相对丰度差异很大,这些结果表明农田土壤中细菌的优势类群相似,但优势类群的相对丰度受土壤类型或质地和种植作物品种的影响[8, 29, 35-36]。施肥和灌溉改变了土壤理化性质,因此对土壤中细菌优势类群相对丰度也产生了一定的影响。灌溉显著改变了放线菌门(<0.001)、拟杆菌门(<0.01)和Saccharibacteria(<0.05)的相对丰度,但施肥没有显著改变各优势类群的相对丰度,这些结果表明本研究中灌溉对土壤细菌群落组成的影响强于肥料类型。放线菌门能够促进土壤中动植物残体的腐烂,同时在土壤氮素循环中也具有一定的作用[36]。王伏伟等[32]研究发现施肥和秸秆还田显著提高了放线菌门的相对丰度,并且土壤全氮含量是提高其相对丰度的重要原因。本研究中,有机肥和化肥处理间放线菌门相对丰度差异不显著,有机肥处理中,灌溉增加了土壤全氮含量却降低了放线菌门的相对丰度。放线菌门相对丰度与全氮含量(=0.047,0.853)无显著相关关系,而与土壤含水量(=-0.799,<0.001)和土壤pH(=-0.570,<0.05)显著相关,表明放线菌门的相对丰度不仅受土壤全氮含量影响,还受到土壤含水量和土壤pH的影响。
前人研究表明施肥[15,28,32]和灌溉[37]等农业管理措施对土壤细菌群落结构有一定影响。本研究发现施肥和灌溉均改变了细菌的群落结构,但不同肥料类型和灌溉次数对细菌群落结构的影响存在差异(图3-a,3-b)。灌溉条件相同的处理具有相似的群落结构,表明灌溉对土壤细菌群落结构的影响强于肥料类型。大量研究也都证实土壤pH、全氮含量和有机碳含量显著影响细菌的群落结构[19,33,38]。本研究中,土壤含水量(<0.001)、土壤pH(<0.001)、全氮含量(<0.01)和有机碳含量(<0.01)与细菌群落结构显著相关,并且土壤含水量与5个主要门类群(变形菌门、放线菌门、拟杆菌门、Saccharibacteria和浮霉菌门)的相对丰度存在显著相关关系,表明土壤含水量是影响细菌群落结构的主要原因。此外,OTU水平上全氮含量与细菌群落结构显著相关,但门水平上全氮含量与主要门类群相对丰度均无显著相关关系,表明全氮含量对细菌不同分类学水平上的群落结构影响存在差异。本研究中,灌溉对细菌群落结构的影响强于施肥,可能是由于灌溉提高了土壤含水量和土壤pH,这两个因素都是影响细菌群落结构的重要因素[6, 16, 38]。
不同施肥和灌溉制度对土壤细菌群落组成和结构都有一定影响,本研究只分析了土壤细菌群落的整体变化,并未对特殊功能类群的微生物进行研究。由于这些特殊功能类群的微生物参与的土壤代谢途径及功能的差异,不同施肥和灌溉制度引起的细菌数量、多样性和群落结构差异是否导致土壤肥力、健康状况和生态系统功能变化有待于进一步研究。
4 结论
4.1 长期不同施肥和灌溉制度对土壤理化性质产生了一定影响。在两种施肥处理中,灌溉均显著提高了土壤含水量和土壤pH,施有机肥比施化肥显著提高了土壤有机碳含量。
4.2 灌溉次数增加显著提高了土壤细菌数量,化肥和有机肥处理中分别提高了1.17—1.60和0.76—1.93倍;而除CI2和MI2外,不同施肥处理间细菌数量差异不显著。灌溉显著改变了细菌群落多样性指数,而施肥对细菌群落多样性指数影响均不显著。
4.3 施肥和灌溉都改变了细菌群落组成和结构,且灌溉的作用强于施肥。此外,土壤含水量和土壤pH与细菌数量、多样性和群落结构均存在显著相关关系,是影响细菌群落变化的主要环境因子。
[1] NANNIPIERI P, ASCHER J, CECCHERINI M T, LANDI L, PIETRAMELLARA G, RENELLA G. Microbial diversity and soil functions., 2003, 54(4): 655-670.
[2] KENNEDY A C, SMITH K L. Soil microbial diversity and the sustainability of agricultural soils., 1995, 170(1): 75-86.
[3] BRUSSAARD L, DE RUITER P C, BROWN G G. Soil biodiversity for agricultural sustainability., 2007, 121(3): 233-244.
[4] 张玉铭, 张佳宝, 胡春胜, 李晓欣, 朱安宁. 华北太行山前平原农田土壤水分动态与氮素的淋溶损失. 土壤学报, 2006, 41(3): 17-25.
ZHANG Y M, ZHANG J B, HU C S, LI X X, ZHU A N. Nitrite leaching in wheat-maize rotation field in the North China Plain., 2006, 41(3): 17-25. (in Chinese)
[5] ABBOTT L K, MURPHY D V.Netherlands: Kluwer Academic Publishers, 2003.
[6] 袁红朝, 秦红灵, 刘守龙, 童成立, 魏文学, 吴金水. 长期施肥对红壤性水稻土细菌群落结构和数量的影响. 中国农业科学, 2011, 44(22): 4610-4617.
YUAN H Z, QIN H L, LIU S L, TONG C L, WEI W X, WU J S. Response of abundance and composition of the bacterial community to long-term fertilization in paddy soils., 2011, 44(22): 4610-4617. (in Chinese)
[7] 乔洁, 毕利东, 张卫建, 沈仁芳, 张斌, 胡锋, 刘艳丽. 长期施用化肥对红壤性水稻土中微生物生物量、活性及群落结构的影响. 土壤, 2007, 39(5): 772-776.
QIAO J, BI L D, ZHANG W J, SHEN R F, ZHANG B, HU F, LIU Y L. Effects of long-term chemical fertilization on soil microbial biomass, activity and community in paddy soil in red soil region of China., 2007, 39(5): 772-776. (in Chinese)
[8] 丁建莉, 姜昕, 关大伟, 马鸣超, 赵百锁, 周宝库, 曹凤明, 李力, 李俊. 东北黑土微生物群落对长期施肥及作物的响应. 中国农业科学, 2016, 49(22): 4408-4418.
DING J L, JIANG X, GUAN D W, MA M C, ZHAO B S, ZHOU B K, CAO F M, LI L, LI J. Responses of micropopulation in black soil of northeast China to long-term fertilization and crops., 2016, 49(22): 4408-4418. (in Chinese)
[9] BERTHRONG T, BUCKLEY D H, DRINKWATER L E. Agricultural management and labile carbon additions affect soil microbial community structure and interact with carbon and nitrogen cycling., 2013, 66(1): 158-170.
[10] WEI D, YANG Q, ZHANG J Z, WANG S, CHEN X, ZHANG X, LI W Q. Bacterial communities structure and diversity in a black soil as affected by long-term fertilization., 2008, 18(5): 582-592.
[11] MA H K, BAI G Y, SUN Y, KOSTENKO O, ZHU X, LIN S, RUAN W B, ZHAO N X, BEZEMER T M. Opposing effects of nitrogen and water addition on soil bacterial and fungal communities in the Inner Mongolia steppe: A field experiment., 2016, 108: 128-135.
[12] PAUL K I, POLGLASE P J, O'CONNELL A M, CARLYLE J C, SMETHURST P J, KHANNA P K. Defining the relation between soil
water content and net nitrogen mineralization., 2003, 54(1): 39-48.
[13] HAN X, WANG R, LIU J, WANG M, ZHOU J, GUO W. Effects of vegetation type on soil microbial community structure and catabolic diversity assessed by polyphasic methods in north China., 2007, 19(10): 1228-1234.
[14] SHOKRALLA S, SPALL J L, GIBSON J F, HAJIBABAEI M. Next-generation sequencing technologies for environmental DNA research., 2012, 21(8): 1794-1805.
[15] 孙瑞波, 郭熙盛, 王道中, 褚海燕. 长期施用化肥及秸秆还田对砂姜黑土细菌群落的影响. 微生物学通报, 2015, 42(10): 2049-2057.
SUN R B, GUO X S, WANG D Z, CHU H Y. The impact of long-term application of chemical fertilizers and straw returning on soil bacterial community., 2015, 42(10): 2049-2057. (in Chinese)
[16] MCHUGH T A, SCHWARTZ E. Changes in plant community composition and reduced precipitation have limited effects on the structure of soil bacterial and fungal communities present in a semiarid grassland., 2015, 388(1/2): 175-186.
[17] SUN M, XIAO T F, NING Z P, XIAO E Z, SUN W M. Microbial community analysis in rice paddy soils irrigated by acid mine drainage contaminated water., 2015, 99(6): 2911-2922.
[18] 钱永, 张兆吉, 费宇红, 陈京生张凤娥, 王昭. 华北平原浅层地下水可持续利用潜力分析. 中国生态农业学报, 2014, 22(8): 890-897.
QIAN Y, ZHANG Z J, FEI Y H, CHEN J S, ZHANG F E, WANG Z. Sustainable exploitable potential of shallow groundwater in the North China Plain., 2014, 22(8): 890-897. (in Chinese)
[19] WU M N, QIN H L, CHEN Z, WU J S, WEI W X. Effect of long-term fertilization on bacterial composition in rice paddy soil., 2011, 47(4): 397-405.
[20] 鲍士旦. 土壤农化分析. 北京: 中国农业出版社, 2000.
BAO S D.Beijing: China Agriculture Press, 2000. (in Chinese)
[21] SUZUKI M T, TAYLOR LT, DELONG E F. Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays., 2000, 66(11): 4605-4614.
[22] WALTER J, TANNOCK G W, TILSALATIMISJARVI A, RODTONG S, LOACH D M, MUNRO K, ALATOSSAVA T. Detection and identification of gastrointestinalspecies by using denaturing gradient gel electrophoresis and species-specific PCR primers., 2000, 66(1): 297-303.
[23] MCBAIN A J, BARTOLO R G, CATRENICH C E, CHARBONNEAU D, LEDDER R G, RICKARD A H, SYMMONS S A, GILBERT P. Microbial characterization of biofilms in domestic drains and the establishment of stable biofilm microcosms., 2003, 69(1): 177-185.
[24] FADROSH DW, MA B, GAJER P, SENGAMALAY N, OTT S, BROTMAN R M, RAVEL J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform., 2014, 2(1): 2-7.
[25] EDGAR R C. UPARSE: highly accurate OTU sequences from microbial amplicon reads., 2013, 10(10): 996-998.
[26] WANG Q, GARRITY G M, TIEDJE J M, COLE J R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy., 2007, 73(16): 5261-5267.
[27] SCHLOSS P D, GEVERS D, WESTCOTT S L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies., 2011, 6(12): 1-14.
[28] SHEN J P, ZHANG L M, GUO J F, RAY J L, HE J Z. Impact of long-term fertilization practices on the abundance and composition of soil bacterial communities in northeast China., 2010, 46(1): 119-124.
[29] CHEN X, ZHANG L M, SHEN J P, XU Z H, HE J Z. Soil type determines the abundance and community structure of ammonia- oxidizing bacteria and archaea in flooded paddy soils., 2010, 10(8): 1510-1516.
[30] 刘方春, 邢尚军, 马海林, 陈波, 丁延芹, 杜秉海. 持续干旱对樱桃根际土壤细菌数量及结构多样性影响. 生态学报, 2014, 34(3): 642-649. LIU F C, XING S J, MA H L, CHEN B, DING Y Q, DU B H. Effects of continuous drought on soil bacteria populations and community diversity in sweet cherry rhizosphere., 2014, 34(3): 642-649. (in Chinese)
[31] 徐永刚, 宇万太, 马强, 周桦. 长期不同施肥制度对潮棕壤微生物生物量碳、氮及细菌群落结构的影响. 应用生态学报, 2010, 21(8): 2078-2085.
XU Y G, YU W T, MA Q, ZHOU H. Effects of long-term fertilizations on microbial biomass C and N and bacterial community structure in an aquic brown soil., 2010, 21(8): 2078-2085. (in Chinese)
[32] 王伏伟, 王晓波, 李金才, 叶爱华, 王妍, 车威, 朱林. 施肥及秸秆还田对砂姜黑土细菌群落的影响. 中国生态农业学报, 2015, 23(10): 1302-1311. WANG F W, WANG X B, LI J C, YE A H, WANG Y, CHE W, ZHU L. Effects of fertilization and straw incorporation on bacterial communities in lime concretion black soil., 2015, 23(10): 1302-1311. (in Chinese)
[33] LIU J J, SUI Y Y, YU Z H, SHI Y, CHU H Y, JIN J, LIU X B, WANG G H. High throughput sequencing analysis of biogeographical distribution of bacterial communities in the black soils of northeast China., 2014, 70: 113-122.
[34] GIRVAN M S, CAMPBEL C D, KILLHAM K, PROSSER J I, GLOVER L A. Bacterial diversity promotes community stability and functional resilience after perturbation., 2005, 7(3): 301-313.
[35] CHU H Y, FIERER N, LAUBER, C L, CAPORASO J G, KNIGHT R, GROGAN P. Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes., 2010, 12(11): 2998-3006.
[36] LAUBER C L, STRIKLAND M S, BRADFORD M A, FIERER N. The influence of soil properties on the structure of bacterial and fungal communities across land-use types., 2008, 40(9): 2407-2415.
[37] 侯海军, 张文钊, 沈建林, 王聪秦红灵. 水分管理对稻田细菌丰度与群落结构的影响. 生态环境学报 2016, 25(9): 1431-1438. HOU H J, ZHANG W Z, SHEN J L, WANG C, QIN H L. Effect of water management on soil bacterial abundance and community in the rice paddy field., 2016, 25(9): 1431-1438. (in Chinese)
[38] ZHOU J, GUAN D, ZHOU B, ZHAO B, MA M, QIN J, JIANG X, CHEN S, CAO F, SHEN D, LI J. Influence of 34-years of fertilization on bacterial communities in an intensively cultivated black soil in northeast China., 2015, 90: 42-51.
(责任编辑 李云霞)
Effects of Long-term Different Fertilization and Irrigation Managementson Soil Bacterial Abundance, Diversity and Composition
YANG YaDong, WANG ZhiMin, ZENG ZhaoHai
(College of Agronomy and Biotechnology, China Agricultural University, Beijing 100193)
【Objective】This study was carried out to investigate the bacterial community in a wheat-maize rotation field during wheat growing season in northern China. The effects of long-term different fertilization and irrigation regimes on the abundance, diversity and composition of bacterial community were identified. It will provide evidences for further optimizing fertilization and irrigation managements, improving soil productivity, and maintaining soil microbial diversity.【Method】Based on a long-term fertilization and irrigation experiment carried out in Wuqiao Experimental Station of China Agricultural University, six different soil samples were collected after wheat was harvested, including chemical fertilization and no irrigation (CI0), chemical fertilization and irrigation at the jointing stage (CI1), chemical fertilization and irrigation at the jointing and filling stages (CI2), manure fertilization and no irrigation (MI0), manure fertilization and irrigation at the jointing stage (MI1), and manure fertilization and irrigation at the jointing and filling stages (MI2). The abundance, diversity and composition of the bacteria community in different soil samples were revealed by using real-time PCR and Illumina Miseq sequencing platform, targeting the 16S rRNA gene. Correlation analysis was carried out between soil properties and the bacteria community abundance, diversity and structure.【Result】Irrigation significantly increased soil moisture and soil pH, and manure fertilization significantly increased Total Organic Carbon (TOC) content compared with no irrigation and chemical fertilization treatments, respectively. The bacterial 16S rRNA gene copy numbers ranged from 4.34×109to 1.39×1010g-1in different treatments. Irrigation significantly increased the bacterial 16S rRNA gene copy number by 1.17-1.60 and 0.76-1.93 times in the chemical and manure fertilization treatments, respectively. The bacterial α diversity index results showed that irrigation but not fertilization significantly affected bacterial α diversity index. At the phylum level, thirty-nine phyla were obtained in all the treatments, among which Proteobacteria, Actinobacteria, Chloroflexi, Acidobacteria and Bacteroidetes were the predominant phyla, accounting for 77.22%-86.28% of the total reads. There were significant variations in relative abundance of Actinobacteria (11.09%-27.01%), Bacteroidetes (5.45%-12.13%) and Saccharibacteria (2.41%-3.77%) among different treatments. Irrigation significantly decreased the relative abundance of Actinobacteria and Saccharibacteria by 36.48%-48.03%, 22.17%-33.67% and 15.21%-45.54%, 13.40%-23.97% in the chemical and manure fertilization treatments, respectively. Hierarchical clustering and principal component analysis (PCA) results showed that both fertilization and irrigation managements affect the bacterial structure, and irrigation had a stronger effect than fertilization on the bacterial structure. In addition, there were significant correlations between soil moisture, soil pH, Total Nitrogen (TN) content, and TOC content and the abundance, α diversity index and structure of the bacterial community.【Conclusion】Irrigation greatly altered the abundance, diversity, and structure of the bacterial community, while fertilization had a minor effect on the abundance and structure of the bacterial community, and soil moisture and soil pH were the potential environmental factors associated with the bacterial community variations.
manure fertilizer; irrigation; bacterial community diversity; high-throughput sequencing; northern China
2017-06-21;
2017-08-25
国家重点研发计划项目(2016YFD0300205-01)、国家自然基金(31671640)、国家公益性行业(农业)科研专项(201503121-11)
杨亚东,E-mail:yadong_tracy@sina.com。
曾昭海,Tel:010-62731211;E-mail:zengzhaohai@cau.edu.cn
猜你喜欢
当代水产(2022年8期)2022-09-20
昆明医科大学学报(2022年2期)2022-03-29
中国麻业科学(2021年5期)2021-12-02
食品安全导刊(2021年20期)2021-08-30
环境卫生工程(2021年1期)2021-03-19
今日农业(2020年19期)2020-12-14
中国化肥信息(2020年6期)2020-11-20
种子(2020年9期)2020-10-22
河南科学(2020年3期)2020-06-02
现代园艺(2020年7期)2020-04-22
