
超高效液相色谱-串联质谱法检测蔬菜中除虫菊素残留
2018-01-25 09:10:13李凌云许晓敏黄晓冬刘广洋郑姝宁徐东辉裴志国
分析测试学报 2018年1期
李凌云,许晓敏,林 桓,钱 洪,黄晓冬,刘广洋,郑姝宁,徐东辉*,裴志国
(1.中国农业科学院蔬菜花卉研究所 农业部蔬菜质量安全控制重点实验室 农业部园艺作物生物学与种质创制重点实验室,北京 100081;2.中国科学院生态环境研究中心 环境化学与生态毒理学国家重点实验室,北京 100085)
除虫菊素是从除虫菊中提取出的植物源性农药,具有高效、广谱、低毒、不易产生抗性等特点,因此被广泛应用于卫生杀虫领域[1-2]。除虫菊素由6个结构极相似的化合物组成[3],即除虫菊素Ⅰ(Pyrethrin Ⅰ)、除虫菊素Ⅱ(Pyrethrin Ⅱ)、瓜叶菊素Ⅰ(Cinerin Ⅰ)、瓜叶菊素Ⅱ(Cinerin Ⅱ)、茉酮菊素Ⅰ(Jasmolin Ⅰ)和茉酮菊素Ⅱ(Jasmolin Ⅱ)。其中除虫菊素Ⅰ和除虫菊素Ⅱ的含量最高,其它4种化合物的含量相对较低。
除虫菊素的残留问题正逐渐引起重视。如我国新版的《食品中农药最大残留限量》GB2763-2016中新增了除虫菊素的残留限量,规定大白菜中除虫菊素的最大残留限量为1.0 mg/kg。但该标准并未给出可以依据或参照的检测方法。另外该标准规定除虫菊素的残留量以除虫菊素Ⅰ和除虫菊素Ⅱ之和计算。然而除虫菊素包括6个组分,对每个组分进行快速准确的定性定量分析,对于真实反映除虫菊素的残留现状,确保人们的食品安全非常重要。
目前报道的除虫菊素残留检测方法有液相色谱法[4]、气相色谱-质谱联用法[5-6]和液相色谱-串联质谱法[7-12],基质主要涉及土壤[7]、茶叶[8]、婴儿食品[9]、食品[5,10-12]等。而采用液相色谱-串联质谱检测蔬菜中除虫菊素残留的研究较少。虽然有些多残留检测对象中包括除虫菊素,但仅涉及除虫菊素的某个成分,并不能满足检测需要。如国标GB/T 20769-2008《水果和蔬菜中450种农药及相关化学品残留量的测定 液相色谱-串联质谱法》中包括除虫菊素,但只有除虫菊素Ⅰ,未包括其他5个组分。
液相色谱-三重四极杆串联质谱技术(HPLC-MS/MS)具有分离效果好、灵敏度高、抗干扰能力强、选择性好等优点,目前已成为农残检测的首选方法[13-15]。本研究利用液相色谱-串联质谱技术,建立了蔬菜样品中除虫菊素6个组分的快速检测方法。蔬菜样品经乙腈提取,提取液无需净化,过滤膜后直接上机测定。该方法简单、快速、回收率高,适用于大批量蔬菜样品的筛查检测。
1 实验部分
1.1 仪器与试剂
LC-30A超高效液相色谱仪和LC-MS/MS-8050三重四极杆质谱仪均购自日本Shimadzu公司。
乙腈、甲醇(HPLC级,美国JT Baker公司);乙酸铵(HPLC级,美国Fluka公司);甲酸(HPLC级,Dikma科技);纯净水(Milli-Q超纯水仪制备);除虫菊素标准品(德国Dr.Ehrenstorfer公司)中各成分含量为:除虫菊素Ⅰ(51.7%)、除虫菊素Ⅱ(33.7%)、瓜叶菊素Ⅰ(4.8%)、瓜叶菊素Ⅱ(4.7%)、茉酮菊素Ⅰ(2.9%)、茉酮菊素Ⅱ(1.8%);其他试剂均为分析纯,购自北京化学试剂公司。
1.2 样品处理
称取10.0 g蔬菜试样于50.0 mL离心管中,加入20.0 mL乙腈,振荡提取30.0 min后,加入5.0 g氯化钠,剧烈振荡1.0 min,以5 000 r/min离心3.0 min后,取上层乙腈相,过0.22 μm滤膜,滤液待测。
1.3 色谱条件
色谱柱:Waters Acquity UPLC BEH HSS C18(100 mm×2.1 mm,1.8 μm);流动相:A为乙腈;B为0.1%甲酸水溶液;梯度洗脱条件:0~10 min,60%~80%A;10~10.1 min,80%~100%A;10.1 ~15 min,100%A;15~15.1 min,100%~60%A;15.1~20 min,60%A;流速:0.3 mL/min;柱温:40 ℃;进样量:1 μL。
1.4 质谱条件
ESI离子源;接口电压4 000 V;正离子扫描;MRM监测模式;雾化气(氮气)流速3 L/min,干燥气(氮气)流速10 L/min,加热气(空气)流速10 L/min;接口温度300 ℃;脱溶剂管温度250 ℃;加热模块温度400 ℃;碰撞气(氩气)压力2.7×105Pa。
2 结果与讨论
2.1 质谱条件的优化
将除虫菊素配制成100 μg/L的标准溶液。优化质谱参数时,将除虫菊素标准溶液在不接色谱柱的情况下直接通过液相色谱注入质谱。以甲醇-水(50∶50,体积比)为流动相时,只能得到除虫菊素Ⅰ和除虫菊素Ⅱ的[M+H]+离子,分别为m/z329.2和373.2,瓜叶菊素Ⅰ、瓜叶菊素Ⅱ、茉酮菊素Ⅰ、茉酮菊素Ⅱ 的母离子不易找到。而以乙腈-水(50∶50)为流动相时,这4个化合物的母离子分别为m/z317.2、361.2、331.2和375.2,且碎片离子相对稳定。在得到6个化合物的母离子[M+H]+后,利用仪器自动优化功能,筛选二级碎片离子,获得碎片离子信息及质谱参数Q1(Quadrupole 1)、Q3(Quadrupole3)和碰撞能量(CE)。优化后的质谱参数见表1。
另外,由于除虫菊素Ⅰ和除虫菊素Ⅱ,瓜叶菊素Ⅰ和瓜叶菊素Ⅱ,茉酮菊素Ⅰ和茉酮菊素Ⅱ每对化合物的结构非常相似,因此它们具有相同的碎片离子。例如除虫菊素Ⅰ和Ⅱ具有相同的碎片离子m/z161和133,瓜叶菊素Ⅰ和Ⅱ的碎片离子为m/z107和77,而茉酮菊素Ⅰ和Ⅱ的碎片离子为m/z163和107。
2.2 流动相的优化
如上所述,相比甲醇-水流动相,6个除虫菊素在乙腈-水作为流动相时的质谱响应更好。因此本实验采用乙腈作为流动相中的有机相。另外考察了在水相中分别添加0.1%甲酸、1.0 mmol/L乙酸铵和0.1%甲酸-1.0 mmol/L乙酸铵对色谱分离及灵敏度的影响,发现3种添加剂对除虫菊素的色谱峰形及分离的影响差别不大,但添加少量的甲酸后,各化合物的离子化效率更高,灵敏度更好,且添加甲酸的效果好于添加乙酸铵。因此本实验最终采用乙腈-0.1%甲酸水溶液作为流动相。
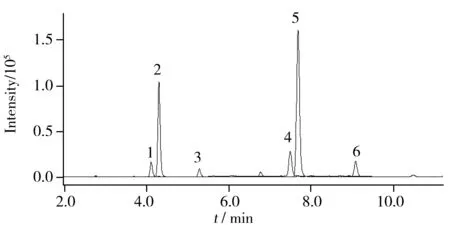
图1 大白菜空白基质中除虫菊素标准溶液的MRM总离子流图(0.1 mg/L)Fig.1 Total ion chromatogram of China cabbage blank matrix spiked with pyrethrins standard solution at 0.1 mg/L in MRM mode1.cinerin Ⅱ;2.pyrethrin Ⅱ;3.jasmolin Ⅱ;4.cinerinⅠ;5.pyrethrinⅠ;6.jasmolinⅠ
在色谱分离时,瓜叶菊素Ⅰ和除虫菊素Ⅰ,瓜叶菊素Ⅱ和除虫菊素Ⅱ的分离比较困难。为此本实验对梯度洗脱条件进行了优化,最终使6个化合物得到完全分离(图1)。6种化合物的保留时间分别为4.110、4.297、5.280、7.488、7.676、9.083 min。
2.3 提取方式的优化
本实验考察了振荡器振荡、均质和超声3种提取方式对除虫菊素提取效率的影响。结果表明,3种提取方式对6种除虫菊素的回收率影响差别不大,回收率分别为91.0%~101.8%、94.3%~104.5%、93.4%~102.4%。由于振荡提取可批量处理样品,节省实验时间,因此本实验采用振荡器振荡提取。
2.4 提取溶剂的选择
本实验分别比较了提取溶剂丙酮、乙酸乙酯、正己烷和乙腈对6种除虫菊素回收率的影响。结果显示,丙酮和乙腈的提取率优于正己烷和乙酸乙酯。以正己烷提取时,6种化合物的回收率为47.4%~103.7%,瓜叶菊素Ⅰ、除虫菊素Ⅰ、茉酮菊素Ⅰ的回收率较低,分别为66.7%、59.1%和47.4%。以乙酸乙酯提取时,6种化合物的回收率为57.3%~81.4%,瓜叶菊素 Ⅱ、除虫菊素Ⅱ、茉酮菊素 Ⅱ的回收率较低,分别为63.3%、63.3%和57.3%。而以丙酮和乙腈提取时,6种化合物的回收率分别为69.8%~97.6%和86.2%~99.5%。相比较而言,乙腈提取得到的结果最好,因此本实验最终选择乙腈作为提取溶剂。
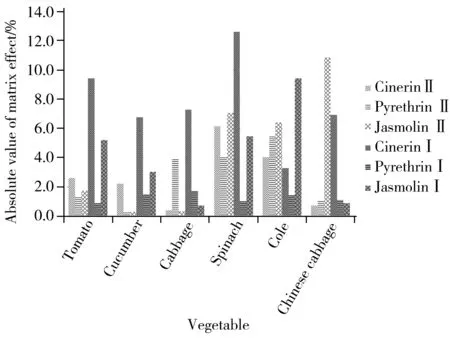
图2 除虫菊素在蔬菜中的基质效应(0.1 mg/L )Fig.2 Matrix effects of pyrethrins at 0.1 mg/L in vegetable matrices
2.5 基质效应考察
基质效应是指基质中的共提取干扰物质影响目标化合物的离子化,从而使目标化合物在仪器上的响应发生增强或抑制的现象。因此在进行农残测定时,对目标化合物在不同基质中的基质效应进行评价是非常重要的。本文采用较为常用的相对响应值法评价基质效应,基质效应=(B/A-1)×100%[16],A为纯溶剂中化合物的响应值,B为蔬菜基质中添加相同浓度农药的响应值。实验考察了0.05、0.1、0.5 mg/L 3个水平下6种化合物在番茄、黄瓜、甘蓝、大白菜、菠菜和油菜6种蔬菜基质中的基质效应。结果表明,0.05 mg/L水平下,基质效应为-11.2%~13.0%;0.1 mg/L水平下,基质效应为-4.1%~12.6%;0.5 mg/L水平下,基质效应为-8.5%~6.0%。3个水平下,6种化合物基质效应的绝对值均小于15%,属于弱基质效应,对测定结果影响较小。图2为除虫菊素在蔬菜中0.1 mg/L水平的基质效应。
2.6 方法的线性关系与定量下限
用菠菜、油菜、甘蓝、番茄、黄瓜和大白菜6种空白基质溶液,准确配制1.0、5.0、10、25、50、100、250、500、1 000 μg/L的系列混合标准工作溶液,按优化实验条件进行LC-MS/MS测定。以峰面积(y)对相应质量浓度(x,μg/L)作标准曲线,得到6种农药的线性方程,各农药的线性范围及相关系数见表1。所有农药的相关系数(r)为0.999 5~0.999 9。其中除虫菊素Ⅰ和除虫菊素Ⅱ的线性范围为1~1 000 μg/L,瓜叶菊素Ⅰ、瓜叶菊素Ⅱ、茉酮菊素Ⅰ和茉酮菊素Ⅱ的线性范围为10~1 000 μg/L。
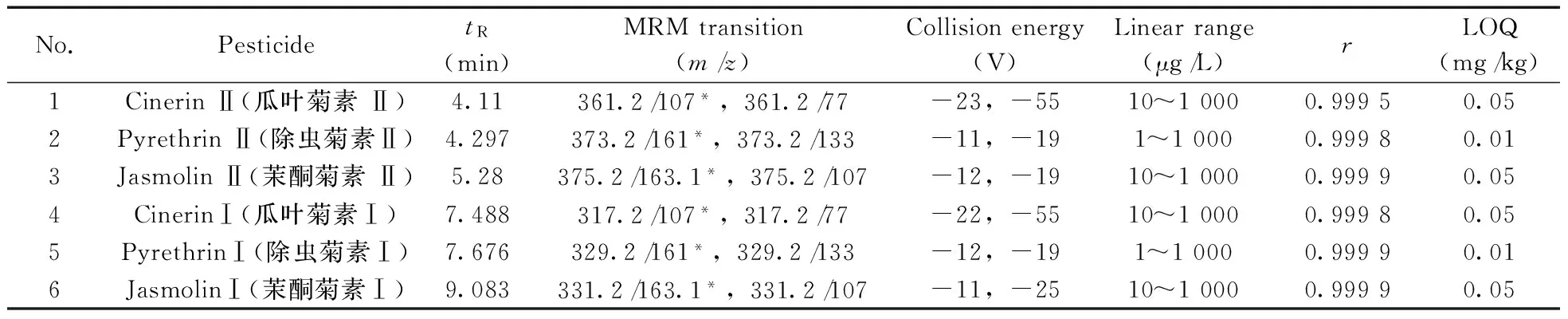
表1 6种化合物的保留时间、MRM离子对、碰撞能量、线性范围、相关系数与定量下限(LOQs)Table 1 Retention times(tR),MRM transitions,collision energies,linear ranges,correlation coefficients and LOQs of six compounds
* quantitative ions
定量下限(LOQ)采用加标回收进行验证,符合一定的准确度和精密度要求的最低加标浓度,确定为定量下限,定量下限是指除虫菊素总量。表1结果显示,除虫菊素Ⅰ和除虫菊素Ⅱ的LOQ为0.01 mg/kg,其他4种化合物的LOQ为0.05 mg/kg。除虫菊素的LOQ低于GB 2763-2016最大残留限量要求。
2.7 方法的回收率与精密度
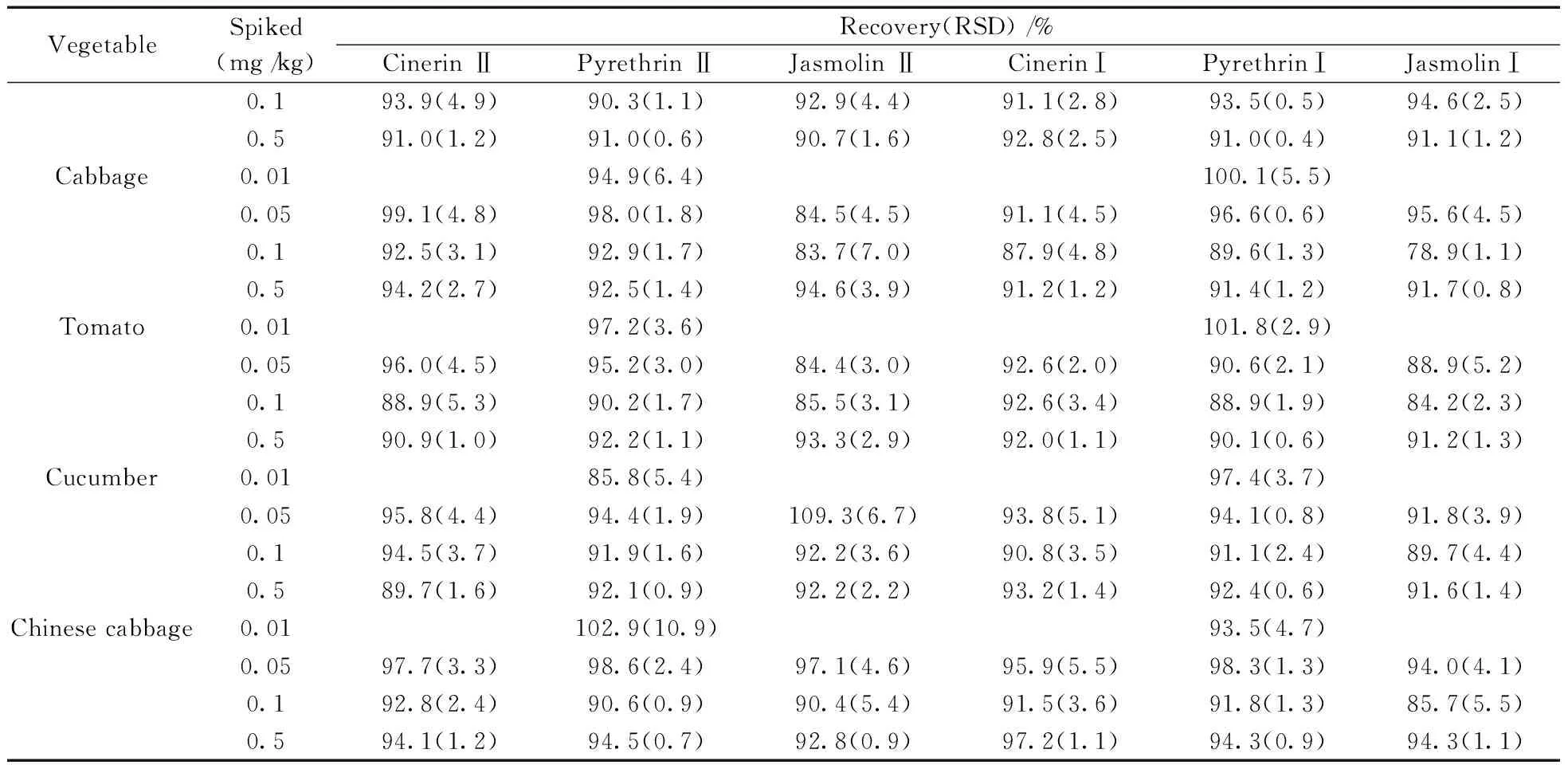
本文采用基质匹配标准溶液外标法定量。在菠菜、油菜、甘蓝、番茄、黄瓜和大白菜6种基质中对除虫菊素进行加标回收率实验,加标水平为0.05、0.1、0.5 mg/kg,每个加标水平重复6次。结果如表2所示,6种物质的平均回收率为73.9%~109.3%,相对标准偏差(RSD)为0.4%~7.7%。方法的准确度和精密度均符合残留分析的要求。另外,由于除虫菊素Ⅰ和除虫菊素Ⅱ的LOQ为0.01 mg/kg,比其它4种化合物的LOQ(0.05 mg/kg)低,因此还对除虫菊素Ⅰ和除虫菊素Ⅱ进行了0.01 mg/kg添加水平的回收率实验,结果见表2。

表2 方法加标回收率和精密度(n=6)Table 2 Spiked recoveries and precisions of the method(n=6)
(续表2)
VegetableSpiked(mg/kg)Recovery(RSD)/%CinerinⅡPyrethrinⅡJasmolinⅡCinerinⅠPyrethrinⅠJasmolinⅠ0.193.9(4.9)90.3(1.1)92.9(4.4)91.1(2.8)93.5(0.5)94.6(2.5)0.591.0(1.2)91.0(0.6)90.7(1.6)92.8(2.5)91.0(0.4)91.1(1.2)Cabbage0.0194.9(6.4)100.1(5.5)0.0599.1(4.8)98.0(1.8)84.5(4.5)91.1(4.5)96.6(0.6)95.6(4.5)0.192.5(3.1)92.9(1.7)83.7(7.0)87.9(4.8)89.6(1.3)78.9(1.1)0.594.2(2.7)92.5(1.4)94.6(3.9)91.2(1.2)91.4(1.2)91.7(0.8)Tomato0.0197.2(3.6)101.8(2.9)0.0596.0(4.5)95.2(3.0)84.4(3.0)92.6(2.0)90.6(2.1)88.9(5.2)0.188.9(5.3)90.2(1.7)85.5(3.1)92.6(3.4)88.9(1.9)84.2(2.3)0.590.9(1.0)92.2(1.1)93.3(2.9)92.0(1.1)90.1(0.6)91.2(1.3)Cucumber0.0185.8(5.4)97.4(3.7)0.0595.8(4.4)94.4(1.9)109.3(6.7)93.8(5.1)94.1(0.8)91.8(3.9)0.194.5(3.7)91.9(1.6)92.2(3.6)90.8(3.5)91.1(2.4)89.7(4.4)0.589.7(1.6)92.1(0.9)92.2(2.2)93.2(1.4)92.4(0.6)91.6(1.4)Chinesecabbage0.01102.9(10.9)93.5(4.7)0.0597.7(3.3)98.6(2.4)97.1(4.6)95.9(5.5)98.3(1.3)94.0(4.1)0.192.8(2.4)90.6(0.9)90.4(5.4)91.5(3.6)91.8(1.3)85.7(5.5)0.594.1(1.2)94.5(0.7)92.8(0.9)97.2(1.1)94.3(0.9)94.3(1.1)
2.8 实际样品的测定
应用所建立的方法对超市和市场上购买的300个菠菜和油菜样品进行检测,结果显示,样品中均未检出除虫菊素残留。
3 结 论
本文采用UPLC-MS/MS技术建立了蔬菜中除虫菊素的快速定性、定量检测方法。样品经乙腈提取,盐析萃取后无需净化,直接上机分析,缩短了样品前处理的时间。该方法简便、快速、灵敏,回收率高,适用于蔬菜样品中除虫菊素的快速检测,并成功应用于实际样品的测定。
[1] Zhang X T,Nie Q L,Gao X.PesticideScienceandAdministration(张夏亭,聂秋林,高欣.农药科学与管理),2003,24(2):22-23.
[2] Nie X Z,Nie R L,Li Z R,Qiu M H.ActaBotanicaYunnanica(聂孝珍,聂瑞麟,李忠荣,邱明华.云南植物研究),1993,15(3):317-319.
[3] Huang X Z.PesticideScienceandAdministration(黄修柱.农药科学与管理),2004,25(4):6-9.
[4] Caboni P,Sarais G,Angioni A,Garau V L,Cabras P.J.Agric.FoodChem.,2005,53:8644-8649.
[5] Rawn D F K,Judge J,Roscoe V.Anal.Bioanal.Chem.,2010,397:2525-2531.
[6] Woudneh M B,Oros D R.J.Chromatogr.A,2006,1135:71-77.
[7] Prestes O D,Padilla-Sánchez J A,Romero-González R,Grio S L,Frenich A G,Martínez-Vida J L.J.Sep.Sci.,2012,35:861-868.
[8] Lu C,Liu X G ,Dong F S,Xua J,Song W C,Zhang C P,Li Y B,Zheng Y Q.Anal.Chim.Acta,2010,678:56-62.
[9] Petrarca M H,Ccanccapa-Cartagena A,Masiá A,Godoy H T,Picó Y.J.Chromatogr.A,2017,1497:28-37.
[10] RuizⅠ,Morales A,Oliva J,Barba A.J.Environ.Sci.Health,2011,46:530-534.
[11] Chung S W C,Lam C H.Anal.Bioanal.Chem.,2012,403:885-896.
[12] Peruga A,Hidalgo L,Sancho J V,Hernández F.J.Chromatogr.A,2013,1307:126-134.
[13] Li L Y,Xu X M,Lin H,Zhao W,Ye R,Huang X D,Zheng S N,Liu X Y,Liu S,Xu D H.Chin.J.Chromatogr.( 李凌云,许晓敏,林桓,赵文,叶融,黄晓冬,郑姝宁,刘新艳,刘肃,徐东辉.色谱),2016,34(9):835-849.
[14] Zhang K,Wong J W,Yang P,Tech Q,DiBenedetto A L,Lee N S,Hayward D J,Makovi C M,Krynitsky A J,Banerjee K,Jao L,Dasgupta S,Simonds R,Schreiber A.J.Agric.FoodChem.,2011,59:7636-7646.
[15] Cao X Y,Pang G F,Jin L H,Kang J,Hu X Y,Chang Q Y,Wang M L,Fan C L.Chin.J.Chromatogr.(曹新悦,庞国芳,金铃和,康健,胡雪艳,常巧英,王明林,范春林.色谱),2015,33(4):389-396.
[16] Li L Y,Wu H,Xu X M,Lin H,Huang X D,Zheng S N,Liu X Y,Xu D H.J.Instrum.Anal.(李凌云,吴华,许晓敏,林桓,黄晓冬,郑姝宁,刘新艳,徐东辉.分析测试学报),2017,36(4):502-506.
猜你喜欢
江西农业学报(2022年8期)2022-11-04 07:37:02
煤化工(2022年3期)2022-07-08 07:24:42
乡村科技(2022年2期)2022-03-25 14:56:16
食品安全导刊(2021年20期)2021-08-30 06:39:48
科学种养(2016年4期)2016-04-19 03:47:43
当代化工研究(2016年5期)2016-03-20 16:21:35
中国资源综合利用(2016年10期)2016-01-22 08:36:09
特产研究(2014年4期)2014-04-10 12:54:22
Sciences in Cold and Arid Regions(2014年6期)2014-03-31 00:28:31
天津化工(2010年5期)2010-09-18 02:55:58
