
桦剥管菌SRAP分子标记反应体系优化与遗传多样性分析
2018-01-25 01:44王秋玉闫绍鹏
中南林业科技大学学报 2018年1期
彭 木,王秋玉,闫绍鹏
(东北林业大学 生命科学学院,黑龙江 哈尔滨 150040)
木材腐朽菌是森林生态系统中森林微生物的重要组成部分,这类菌广泛地生长在各种树木的活立木、枯立木、倒木、伐桩、原木、枕木、板材及许多建筑用材上,因能引起木材腐朽而降低了木材的实用价值[1]。桦剥管菌Piptoporus betulinus又称桦孔菌、桦滴孔菌,是桦木属树木专性木腐菌,造成木材褐色腐朽,在桦树天然林中较为常见。此外,该真菌在民间医药中被当作一种常见的抗寄生虫和抗菌剂,用于伤口和直肠癌等疾病的治疗,是一种非常有前途的医药资源。
近年来,褐腐菌因其强大的降解纤维素能力和独特的降解方式而备受关注,已成为国际上研究的热点。利用其对木质纤维素进行生物预处理,能高效地生产乙醇燃料,同时能作为生物催化剂、发酵碳源等用途,具有广阔的应用前景。目前,有关褐腐菌的报道主要集中在降解木质纤维素、生物质能转换、木材相关降解酶的分泌与木材降解机制等方面的研究。Deswal等[2]研究证明经过预处理的褐腐菌在降解木材水平上比使用纤维素酶处理效果强3倍。Fissore等[3]在木质素预处理过的新西兰辐射松中接种密粘褶菌,结果检测到高达70%的葡萄糖含量。Hastrup等[4]对4种不同的褐腐菌在降解过程中的草酸积累量进行了定量测定,结果发现草酸的含量与木材降解过程中质量减少有关。
相关序列扩增多态性(Sequence-related Ampli fi ed Polymorphism,SRAP)于2001年由Li和Quiros[5]首次提出,是一种以全基因组为靶标,检测开放阅读框及其上游启动子或内含子之间序列多态性的一种分子标记技术。SRAP被认为具有简单高效、重复性好、稳定性高、引物通用性好等特点,且通过正反向引物随机搭配组合,提高了引物的使用效率,也降低了实验成本,因而被广泛地应用于遗传多样性、物种鉴定、遗传连锁图谱构建以及杂种优势预测等方面的研究中[6-8]。Sun等[9]运用SRAP对31个在形态学上具有代表性的灵芝和一些至今没有归类的灵芝菌株进行遗传多样性分析,结果揭示了不同采集地的灵芝菌株遗传多样性与其地理环境之间的相关性。由于毛木耳真菌种质指纹图谱的鉴定还不够完善,Yu等[10]利用ISSR和SRAP技术对19个毛木耳真菌进行了遗传多样性的测定,结果表明SRAP标记相对ISSR具有更加高效性和可取性。Yin等[11]采用SRAP、RAPD和ISSR技术分析了15个不同采集地的凤尾菇菌株,结果表明3种标记对菌株的分类基本上一致。
目前,对于不同采集地桦剥管菌的遗传多样性研究还未见报道。因此,本研究以采自中国东北主要林区——塔河、带岭、帽儿山和敦化4个地点的桦剥管菌菌株为研究材料,利用SRAP技术对其种群遗传多样性进行分析和评价,为桦剥管菌新品种的选育和鉴定以及进一步利用提供理论依据。
1 材料与方法
1.1 试验材料
供试验材料采自4个不同的地点,包括带岭(小兴安岭)、塔河(大兴安岭)、帽儿山(张广才岭)和敦化(长白山)(见表1)。

表1 桦剥管菌采集地信息Table 1 The information of sampling site of P. betulinus
1.2 方 法
1.2.1 桦剥管菌菌株的纯化及DNA的提取
采用訾晓雪等[12]的菌株纯化分离法,对采集的菌株子实体进行分离纯化。采用改良的CTAB法[13]提取桦剥管菌总DNA,并用1%琼脂糖凝胶电泳和分光光度计检测其纯度和浓度,并将DNA样品稀释至10 ng/μL,-20℃保存备用。对提取的15个桦剥管菌DNA样品进行等量混合成基因池,保证混合基因池中DNA的终浓度为10 ng/μL。
1.2.2 PCR反应条件的优化
以吕世翔[14]、曹宇[15]、王玉英等[16]对白腐菌遗传多样性分析中所采用的PCR反应体系的研究结果为基准反应,对SRAP反应体系先进行单因素优化,以找到适合于桦剥管菌的最优反应体系。
(1)基准反应体系
10×Buffer 2.0 μL,Primer 各 15 μmol/L,dNTP 6 mmol/L,Taq polymerase 1 U,DNA 10 ng,Mg2+50 mmol/L,总体积 20 μL。
(2)PCR反应体系的单因素优化
PCR反应体系中,dNTP浓度、Mg2+浓度、DNA模板浓度、引物浓度、Taq polymerase浓度的单因素优化见表2。
(3)SRAP-PCR反应条件
PCR反应条件:94℃ 5 min;94℃ 1 min,35℃ 1 min,72℃ 1 min,5 个循环;94℃ 1 min,56℃ 1 min,72℃ 1 min,35个循环;72℃10 min;4℃保存。PCR扩增产物通过2%琼脂糖凝胶电泳检测。
(4)PCR反应体系的均匀设计优化体系
在PCR反应体系中各因子的单因素优化的基础上,确定各个单因素的最适浓度,并进行均匀设计U20(54)分析最佳的反应体系,均匀设计见表3。根据均匀设计表中的浓度进行PCR,并用Gel-Pro软件分析各泳道中各条带的积分光密度值(Integrating Optical Density,IOD),以确定SRAP-PCR反应体系的最佳体系。

表2 PCR反应体系的单因素优化Table 2 The PCR reaction system by single factor optimization

表3 PCR反应体系均匀设计U20(54)Table 3 PCR reaction system by uniform design method U20(54)
1.2.3 引物设计及筛选
SRAP引物序列参照文献[17],上游引物为me1~me8,下游引物为em1~em11,各引物序列信息见表4。对本试验的所有引物先进行初步筛选,以能扩增出条带数多、重复性好、稳定性高的引物进行后续遗传多样性分析。

表4 TRAP和SRAP分子标记技术的引物序列Table 4 The sequences of primers used in two kinds of molecular markers
1.3 统计分析
根据PCR结果所得的凝胶电泳图谱,将不同地点菌株DNA的电泳谱带按照相同迁移位置上有条带的记为“1”,没有条带的记为“0”。为了评估桦剥管菌的遗传多样性,利用POPGENE version 1.31软件分析了其遗传多样性参数[18],包括多态性条带数、多态性条带比率、观测等位基因数(Na)、有效等位基因数(Ne)、Nei’s遗传多样性(H)、Shannon指数(I)、遗传一致度、基因流(Nm)、Nei’s遗传分化(Gst);利用NTSYS-pc软件,运用非加权组平均法(UPGMA)进行系统聚类分析。
2 结果与分析
2.1 反应体系的优化结果
2.1.1 dNTP浓度的优化结果
不同浓度下PCR结果见图1-A。由图1-A可知,当dNTP的浓度较低时,条带模糊,呈弥散状;随着dNTP浓度的增加,条带变得清晰,并在某一浓度范围内(泳道10、11、12),条带最为清晰、整体,但是超过这一范围后,扩增条带数明显减少至完全没有条带,说明PCR反应中并不是dNTP浓度越高越好。同时,鉴于节约原则,后续实验选择以效果最好、用量最少的dNTP为宜,故后续实验选择10 mmol/L(泳道10)进行PCR扩增。
2.1.2 Mg2+浓度的优化结果
不同Mg2+浓度下PCR结果见图1-B。在低浓度(泳道1~10)时,扩增结果没有条带;随着浓度的不断增加(泳道11~15),条带数增多,条带清晰;但超过一定浓度时,扩增又受到限制。因此,在本试验中选择了Mg2+浓度60 mmol/L作为PCR的反应浓度,此时所获得的扩增产物较为理想。

图1 不同因素对PCR反应结果的影响Fig. 1 PCR results of different factor’s concentration
2.1.3 DNA模板浓度的优化结果
本实验中,共设计20个模板梯度,不同DNA浓度对PCR扩增结果见图1-C。当模板浓度较高或较低时,扩增得到的条带数较少;当模板浓度处于2~10 ng时,条带数逐渐清晰,拖尾消失;当浓度超过10 ng时,又有少许拖尾现象。因此,后续实验选择DNA模板浓度为10 ng。
2.1.4 引物浓度的优化结果
不同引物浓度下PCR结果见图1-D。由图1-D可知,从泳道1到10,条带逐渐增加,且清晰;从泳道11到20,条带又逐渐减少且模糊。相比于泳道10和11,泳道11能明显看到条带拖尾,不便于统计分析,因此,反应体系中引物的加入以10 ng为宜,能得到较多的条带数,而且清晰便于统计。
2.1.5 Taq polymerase浓度的优化结果
从图1-E上可以看出,使用20个不同浓度的Taq polymerase,PCR扩增产物条带数基本上一致,变化不大。因此,为了节约酶量,后续实验的Taq polymerase浓度为1 U。
2.1.6 均匀设计优化PCR反应体系结果
大量研究证实,均匀设计实验是一种较好的实验条件优化方法,与单因素试验和正交设计相比,均匀设计能从全部试验点中挑选出部分代表性的试验点,而这些试验点能准确地反映出体系的主要特征,从而大大减少试验次数[19-20]。本实验用均匀设计法扩增结果的电泳图见图2。用软件Gel-Pro对各泳道的条带数进行统计,并计算其总IOD值,结果见图2和表5。由表5可知,条带最多为16条(16、2泳道)、15条(1、6泳道)、14条(4、11、12、13、17、18泳 道);泳道17、1、16中条带最多的总IOD分别是2 589 943.5、2 350 985.4、2 347 941.4。综合以上结果,确定最佳的PCR反应体系为第17号反应管中的体系,即为:10×Buffer 2.0 μL、Primer 各8 μmol/L、dNTP 16 mmol/L、Taq polymerase 2.5 U、DNA 12 ng、Mg2+45 mmol/L,总体积 20 μL。
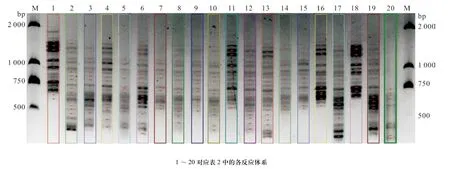
图2 Gel-Pro软件划分各泳道及条带(黑点代表条带)Fig. 2 The band lane and number analyzed using Gel-Pro software (red dot represents band)
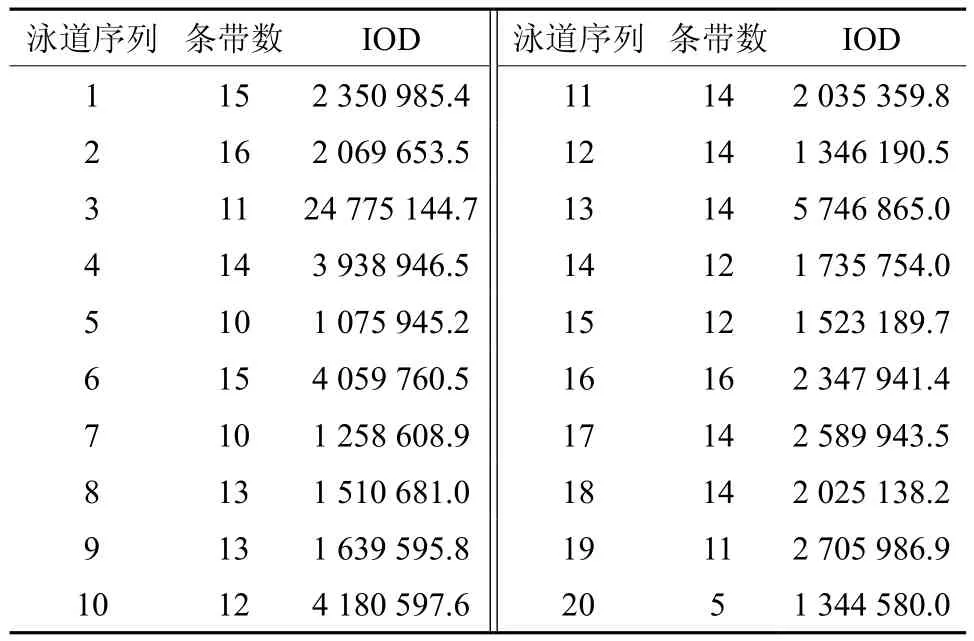
表5 Gel-Pro软件分析各泳道中条带的总积分光密度值(IOD)Table 5 The IOD values in each band using Gel-Pro software
2.2 基于SRAP的遗传多样性分析
从88对SRAP引物组合中,选择了15对扩增产物条带重复性好、稳定性高的引物对所有样品进行多态性分析(见图3)。对15个菌株进行SRAP遗传多样性分析,结果扩增的片段大小分布在100~2 000 bp之间,共扩增出239条清晰的条带,其中237条为多态性条带,占总条带数的99.16%(见表6),平均每对引物产生15.8条多态性条带,说明SRAP技术在15个样品中的多态性水平较高。此外,不同引物扩增的条带数不同,扩增条带数从10~22条不等,且基本为多态性条带,反映了4个地点的桦剥管菌之间具有较高的基因多样性水平。

表6 SRAP引物扩增的条带数统计Table 6 The PCR products of each primers in P. betulinus based on SRAP marker
不同地点间桦剥管菌的SRAP遗传多样性指数值在帽儿山地区中最高,在敦化地区最低(见表7),说明带岭的菌株拥有相对较高的遗传变异。15对引物在4个地点间扩增出的多态位点为44~150,多态位点比例为18.41%~62.76%;等位基因观察值(Na)、有效等位基因平均数(Ne)、Nei’s多样性指数、Shannon多样性指数分别为1.405 9~1.627 6、1.130 2~1.409 8、0.076 3~0.234 7、0.111 3~0.347 8(见表7)。基于SRAP分子标记,4个采集地的桦剥管菌群体总基因多样性(Ht)为0.287 7,群体内基因多样性(Hs)为0.170 3,群体间遗传分化系数(Gst)为0.408 3,各群体间基因流(Nm)为0.724 7(见表8)。

图3 引物me7/me10对15个桦剥管菌菌株的SRAP扩增结果Fig.3 SRAP ampli fi ed pro fi les by primers me7/me10 of 15 P. betulinus strains
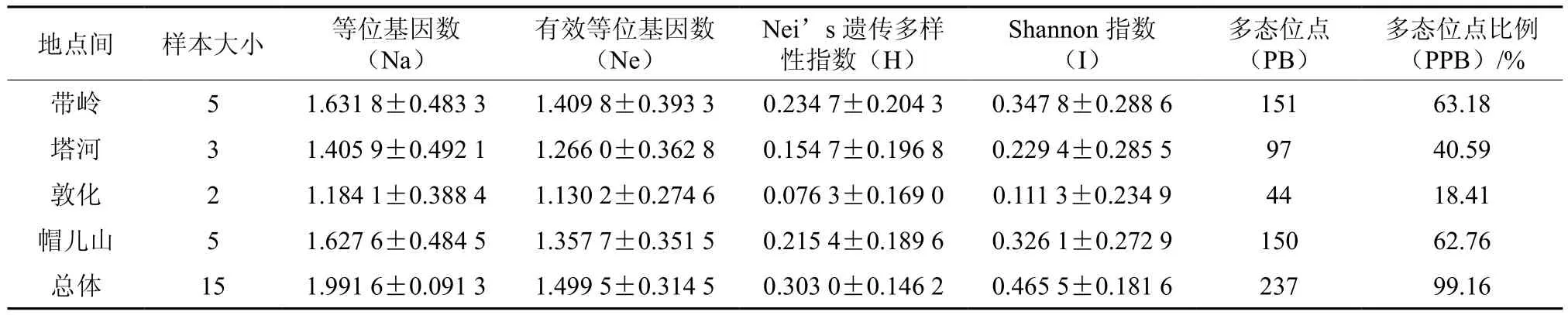
表7 不同桦剥管菌地点间的遗传多样性指数比较Table 7 Nei’s analysis of gene diversity in P. betulinus from different collections

表8 桦剥管菌的Nei’s遗传多样性指数Table 8 Nei’s analysis of gene diversity in P. betulinus populations
不同地点间桦剥管菌的遗传距离和遗传一致度见表9。由表9可知,带岭与塔河、带岭与敦化的遗传距离和遗传相似系数分别最近(0.175 3,0.839 2)和最远(0.246 3,0.781 7),这和地理距离基本一致,表明地理距离越近,亲缘关系越近。此外,对桦剥管菌种群内和种群间的分子变异情况进行分析,结果(见表10)表明,88.08%的变异率来源于种群内,而种群间的变异率仅为11.92%,这意味着桦剥管菌的遗传分化主要发生在种群内,不同的种群间由于地理隔离等因素影响了遗传分化的结果,这与SRAP的群体间遗传分化系数(Gst)结果相一致。
利用NTSYS 2.1软件对15个桦剥管菌菌株进行系统聚类分析,结果见图4。由图4可知,遗传相似性系数在0.56~0.86之间。15个菌株被明显地分成了两大组(Group I和Group II),Group I组包括带岭和塔河的全部菌株,说明带岭和塔河的菌株的遗传关系较近,遗传组成相似;Group II组包括帽儿山和敦化的全部菌株。总体而言,基本上符合样品采集地间的距离远近关系,表明在同一地域的菌株其遗传背景较为相似,很好地体现了桦剥管菌菌株间遗传特性与地理分布的特点,即Group I组主要来自松嫩平原地区,该地区具有较低的降水量,而Group II组主要来自辽东半岛,具有相对较高的降水量和高海拔山脉。

表9 不同桦剥管菌地点间的遗传距离(下三角)和遗传一致度(上三角)Table 9 Nei’s genetic distance and genetic identity coefficient of groups of P. betulinus from different collections
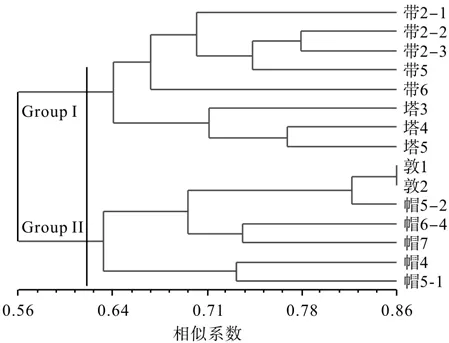
图4 基于SRAP分子标记的遗传相似性系数对15个桦剥管菌菌株进行非加权组平均法系统的聚类结果Fig.4 UPGMA dendrogram of fi fteen P. betulinus strains based on Jaccard’s coef fi cient of SRAP markers
3 结论与讨论
本研究首次对来自东北主要林区不同种群的桦剥管菌进行遗传多样性分析,为桦剥管菌多态性鉴定以及进一步利用提供理论依据,结果表明桦剥管菌菌株间遗传特性与地理分布间的近缘关系。目前,关于真菌种质资源的研究仅限于某些常见的食用和药用真菌,而对木材腐朽菌,如桦剥管菌鲜有报道。较多有关丛枝菌根(AM)真菌的种质资源的报道,研究表明AM真菌种质资源丰富,生态适应性强,宿主范围十分广泛,但其在不同生态系统中的分布有着明显的差异[21],同时菌种鉴定的难度限制了AM真菌的进一步研究。中国幅员辽阔,自然条件相当优越,真菌资源极为丰富,因此,需要投入更多的人力和物力以丰富真菌的菌种数据库。因此,本试验材料采自东北主要林区,目的是考察桦剥管菌种质资源分布,并研究它们的遗传多样性,这将为桦剥管菌开发和应用奠定理论基础。
目前对于各种PCR反应体系的优化有着多种方法,包括单因子试验、正交设计和均匀设计等方法[22-23]。单因子通过各因素的组合找出最适的条件,但是忽略了各因素之间的综合作用[24];正交设计解决了混合离散变量的优化问题[25],而均匀设计舍弃了正交设计中的整齐可比性,让试验点得到均匀分散,具有很好的代表性,同时减少了实验次数[26]。因此,在本实验中,先通过单因子试验确定各因素的最佳浓度,然后通过均匀设计进一步确定各因素之间的最适组合,保证了实验的准确有效性。通过优化后的结果可以看出,最优体系扩增的条带数明显的增多,总条带数也相应的增多,这增加了多态性条带的比例概率,在进行种群遗传多样性分析时,更能体现种群的多样性水平。
本试验共筛选到15对SRAP引物,对来自4个采集地点的桦剥管菌株进行遗传多样性分析,结果共扩增出239条清晰的条带,其中237条为多态性条带,占总条带数的99.16%,高于吕世翔[14]采用TRAP引物对3种木腐菌(桦剥管菌、木蹄层孔菌和彩绒革盖菌)扩增得到的77.74%多态性。此外,Ferriol等[27]报道了SRAP技术可提供更多的信息量,且其与形态变异和突变表型间的相关性高于AFLP技术,因此,推断SRAP技术检测物种遗传多样性水平相对丰富的原因可能是SRAP引物的独特特点。同时,聚类结果表明在同一地域的菌株的遗传背景相似,遗传组成相近的特点。这种聚类特点在其他菌株上也有报道,傅常娥[28]运用SRAP技术比较了来自中国、芬兰、俄罗斯、日本和朝鲜等不同国家及地区的20个桦褐孔菌菌株的遗传多样性,聚类分析结果将20个菌株归属6个地点,显示出菌株间的遗传差异与地理分布有较强的相关性。此外,Ma等[29]也采用SRAP标记技术分析松茸的遗传多样性及其遗传关系,结果表明12对SRAP引物在来自13个中国东北不同地区的129个菌株中检测到了丰富的遗传多样性,聚类结果表明遗传距离和地理距离之间存在显著的正相关,而遗传距离与采集地海拔高度之间没有明显的相关性,这与本试验结果相一致。本研究中在4个地点共采集到15个菌株,样本数相对较少,但研究结果也能说明遗传多样性水平高的特点,后续的研究方向将集中在中国东北地区与南方地区的桦剥管菌进行比较,为桦剥管菌的酶学研究奠定提供基础。
[1]杨 晨, 刘 勇, 陈 晓, 等. 油松人工林下真菌群落对凋落物分解的影响[J]. 中南林业科技大学学报, 2016, 36(7):41-47.
[2]Deswal D, Gupta R, Nandal P,et al. Fungal pretreatment improves amenability of lignocellulosic material for its saccharification to sugars [J]. Carbohydrate Polymers, 2014,99(1): 264-269.
[3]Fissore A, Carrasco L, Reyes P,et al.Evaluation of a combined brown rot decay-chemical delignification process as a pretreatment for bioethanol production fromPinus radiatawood chips [J]. Journal of Industrial Microbiology & Biotechnology,2010, 37(9): 893-900.
[4]Hastrup A, Iii F, Lebow P,et al.Enzymatic oxalic acid regulation correlated with wood degradation in four brown-rot fungi [J].International Biodeterioration & Biodegradation, 2012, 75(3):109-114.
[5]Li G, Quiros C. Sequence-related amplified polymorphism(SRAP), a new marker system based on a simple pcr reaction:is application to mapping and gene tagging in Brassica [J].Theoretical Applied Genetics, 2001, 103(2-3): 455-461.
[6]Lin X, Lou Y, Zhang Y,et al.Identi fi cation of Genetic Diversity Among Cultivars of Phyllostachys violascens Using ISSR, SRAP and AFLP Markers [J]. Botanical Review, 2011, 77(3): 223-232.
[7]Lu J, Zhao H, Suo N,et al. Genetic linkage maps of Dendrobium moniliforme and D. of fi cinale based on EST-SSR, SRAP, ISSR and RAPD markers [J]. Scientia Horticulturae, 2012,137(4):1-10.
[8]逯晓萍, 刘丹丹, 王树彦, 等.高丹草遗传效应与杂种表现预测模型 [J]. 作物学报, 2014,40(3):466-475.
[9]Sun S, Gao W, Lin S,et al.Analysis of genetic diversity in Ganoderma population with a novel molecular marker SRAP [J].Applied Microbiology & Biotechnology, 2006, 72(3): 537-543.
[10]Yu M, Ma B, Luo X,et al. Molecular diversity of Auricularia polytricha revealed by inter-simple sequence repeat and sequence-related amplified polymorphism markers [J]. Current Microbiology, 2008, 56(3): 240-245.
[11]Yin Y, Liu Y, Li H,et al.Genetic diversity of Pleurotus pulmonarius revealed by RAPD, ISSR, and SRAP fi ngerprinting[J]. Current Microbiology, 2014, 68(3): 397-403.
[12]訾晓雪, 曹宇, 闫绍鹏, 等. 3种白腐菌木质纤维素酶活性及酶相关基因的TRAP标记遗传多态性[J].林业科学, 2015,(6): 111-118.
[13]王桂娥, 晁群芳, 梁建芳, 等. 改良CTAB法提取新疆一枝蒿干叶基因组DNA [J]. 中国实验方剂学杂志,2015,21(12):19-22.
[14]吕世翔. 三种木腐菌木材降解相关酶以及相关基因TRAP标记的变异[D]. 哈尔滨:东北林业大学, 2010.
[15]曹 宇. 海绵状白腐菌木质纤维素酶活性及其基因的遗传变异研究[D].哈尔滨: 东北林业大学, 2013.
[16]王玉英, 吕世翔, 王秋玉. 3种木腐菌木质纤维素相关酶基因的TRAP标记体系优化与遗传变异初探 [J]. 林业科技,2012,37(2): 17-20.
[17]盛文涛, 周劲松, 汤泳萍, 等. 芦笋SRAP反应体系优化及引物筛选 [J]. 分子植物育种, 2010, 8(3):612-618.
[18]王成树, 李增智. 分子数据的遗传多样性分析方法 [J]. 安徽农业大学学报, 2002,29(1):90-94.
[19]王 兵, 王晓春. 均匀设计直观分析法优化PCR条件 [J]. 检验医学, 2007, 22(5):620-622.
[20]方开泰. 均匀设计及其应用(Ⅲ) [J]. 数理统计与管理,1994(3): 52-55.
[21]常双双, 王承南, 王 森, 等. 5种丛枝菌根真菌对君迁子幼苗光合生长的影响[J].经济林研究, 2016, 34(2):79-85.
[22]余贵湘, 段忠俊, 卢 靖, 等. 施肥和覆盖对澳洲坚果344和OC生长和产量的影响[J].经济林研究, 2016, 34(4):73-79.
[23]牛广俊, 陈清英, 朱 思, 等. 用层次分析法多指标评价优选金花茶超声提取工艺[J].经济林研究, 2015, 33(1):119-122.
[24]胡 薇, 黄儒珠. 均匀设计优化建兰ISSR-PCR体系[J].生命科学研究, 2007, 11(1):58-63.
[25]孙晋科. 巴旦杏RAPD-PCR反应体系的正交优化[J].新疆农业科学, 2007, 44(5):715-719.
[26]郭丽琴, 卫尊征, 张金凤, 等. 均匀设计优化杨属的SRAP—PCR反应体系[J]. 北京林业大学学报, 2010,32(2):34-38.
[27]Ferriol M, PicóB, Nuez F. Genetic diversity of a germplasm collection of Cucurbita pepo using SRAP and AFLP marker [J].Theoretical and Applied Genetics, 2003, 107(2): 271-282.
[28]傅常娥. 桦褐孔菌菌株遗传多样性的SRAP和RSAP分析[D].延边:延边大学, 2014.
[29]Ma D, Yang G, Mu L,et al.Application of SRAP in the genetic diversity of Tricholoma matsutake in northeastern China [J].African Journal of Biotechnology, 2010, 38(9): 6244-6250.
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
计算机应用与软件(2022年6期)2022-07-12
自然灾害学报(2022年2期)2022-05-10
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2021年6期)2021-07-31
昆明医科大学学报(2021年3期)2021-07-22
中国学校体育(2021年10期)2021-04-26
数学大王·低年级(2020年8期)2020-08-14
小雪花·初中高分作文(2016年9期)2016-05-14
实验流体力学(2014年6期)2014-07-10
