
高性能锂硫电池研究进展
2018-01-15 02:04李路路胡南滔魏良明
物理化学学报 2017年12期
刘 帅 姚 路 章 琴 李路路 胡南滔 魏良明 魏 浩
高性能锂硫电池研究进展
刘 帅 姚 路 章 琴 李路路 胡南滔 魏良明 魏 浩*
(上海交通大学电子信息与电气工程学院,薄膜与微细技术教育部重点实验室,上海 200240)
锂硫电池具有理论比容量高(1675 mAh∙g−1)、能量密度高(2600 Wh∙kg−1)、环境友好、价格低廉等性质,是一种高性能的新型储能电池。这些性能使其在电动汽车和便携式设备领域具有重要意义。然而,快速的容量衰减以及较差的循环性能,使锂硫电池还达不到商业应用的要求。本文全面总结了锂硫电池的最新研究进展,详细阐述了锂硫电池的正极、电解质、隔膜以及负极保护,分析了现有锂硫电池存在的缺陷和问题。最后,对锂硫电池未来的发展方向进行了展望。
锂硫电池;正极;负极保护;隔膜;应用
1 引 言
当今人们非常重视可持续能源的开发,减少对环境造成污染。为了实现这一目标,需要减少对化石燃料的使用,并努力向清洁可再生能源转变,如太阳能和风能。然而,可再生能源本身的间断性质使其必须和先进的能量存储系统配套,这种能量储存系统应当能够有效收集可再生能源并且按需释放能量。电池系统作为储存和释放能量的有效中介,在这些领域可以发挥关键作用。通常用于小型便携式电子设备的锂离子电池不能满足固定电网能量存储的高能耗需求1–4。锂离子电池低的能量密度也阻碍了它们在新兴移动运输设备中的应用,如电动汽车等领域。电动汽车与燃油汽车相比,对环境更加友好并且更加安全,这促使研究人员开发出更加合理有效电池能源系统。
同锂离子电池相比,锂硫电池的研究引起了广泛的关注5–11。锂硫电池和锂离子电池的电化学反应机理不同。锂离子电池中锂离子嵌入层状电极材料(例如石墨阳极和锂金属氧化物阴极)中,往往只能嵌入到某些特定位点,能量密度通常为420 Wh∙kg−1。而锂硫电池不仅拥有高的理论比容量(1675 mAh∙g−1)和能量密度(2600 Wh∙kg−1),并且硫元素便宜易得,这些优点使得锂硫电池成为有吸引力的下一代低成本能量存储技术之一。
锂硫电池主要由正极、电解质、隔膜和负极等组成。其正极材料硫元素是由八个硫原子组成的冠状结构,具有非常稳定的热力学性能12,13。硫元素高的充放电性能与S8分子中硫硫键的断裂和重组有关。但目前锂硫电池存在着自身容量衰减快、硫正极电导率低、多硫化物“穿梭效应”、锂离子沉积以及在充放电过程中由于体积变化造成结构改变等问题,使锂硫电池难以投入大规模的商业化生产。
本文主要评述锂硫电池的最新研究进展,从电化学原理入手,详细分析锂硫电池目前的研究成果,并针对锂硫电池的未来发展方向提出了一些建议和展望。

2 电化学原理及面临的挑战
锂硫电池的电化学反应原理:
S8+ 16Li2→ 8Li2S
其中硫具有高理论比容量(1673 mAh∙g−1)。在放电过程中,锂金属阳极(负极)氧化形成锂离子和电子,它们分别通过电解质和外部电路到达硫阴极(正极)。在正极处,硫与锂离子以及电子进行还原反应形成硫化锂。充电过程与之相反。
放电过程:
正极:S8+ 16Li++ 16e−→ 8Li2S
负极:Li → Li++ e−
充电过程:
负极:Li++ e−→ Li
正极:8Li2S → S8+ 16Li++ 16e−
虽然书面化学反应式看似简单,但实际的充/放电反应过程却比较复杂。放电过程中,硫首先锂化形成一系列中间的长链多硫化锂(S8→ Li2S8→ Li2S6→ Li2S4),这种长链物质容易溶解在醚基电解质中,这个过程发生反应的硫占总容量的25% (418 mAh∙g−1)。在进一步锂化时,溶解的长链多硫化物形成短链硫化物物质(Li2S4→ Li2S2→ Li2S),生成固体物质沉淀到电极上,该过程反应硫占总容量的75% (1255 mAh∙g−1)。充电反应过程与放电反应过程正好相反。整体来看,随着反应进行,锂硫电池经历固-液-固体转变,这和传统的锂离子电池的反应机理不同。
阻碍锂硫电池投入实际应用有多方面的原因,主要在于正极和负极。在硫正极,主要遇到以下困难:
(a) 中间体多硫化物溶解到电解质中。在循环过程中,中间长链多硫化物(Li2S4至Li2S8)容易溶解于醚基电解质中,这导致活性材料连续损失到电解质中,部分活性物质会始终保持溶解状态,在放电结束时作为硫化锂到达正极。
(b) 硫和硫化锂的低电导率。硫和硫化锂的电绝缘性和离子传导的绝缘性质导致活性材料得不到充分循环,在放电期间绝缘硫化锂的沉淀也导致阴极表面钝化。
(c) 锂化时硫的体积改变。由于硫和硫化锂之间的密度差(分别2.03和1.66 g∙cm−3),硫在完全锂化为硫化锂时有较大的体积膨胀率,这可能导致电极的破裂和损坏。
在锂负极,主要面临以下困难:
(a) 多硫化物穿梭效应。溶解到电解质中的长链多硫化锂可以到达锂负极,以化学方式还原,并形成低价态化合物,然后这些低价态化合物能够再次回到硫正极被再一次氧化。这种多硫化物穿梭效应发生在电池内部,在循环期间导致自放电,造成较低的库仑效率。
(b) 不均匀的固体电解质中间相(SEI)。锂金属能够在和电解质的界面上和电解质发生反应并在表面上形成SEI层,这种SEI层对离子导电但是对电子绝缘。大多数情况下SEI是不均匀的,不能充分钝化锂金属表面,这将导致锂金属和电解质之间发生副反应,消耗锂金属和电解质,导致电池的可逆性变差,造成较低的库仑效率。
(c) 锂金属的枝晶生长。锂枝晶的生长导致SEI的破裂,进一步消耗锂金属和电解质,导致电池电解质的不断耗尽。并且较厚SEI层会导致较高阻抗,影响电池的循环效率。
针对以上问题,研究人员提出了各种解决方案,其中最常见的是对正极材料改性封装。经常用的改性封装材料是碳材料,然而,碳材料和多硫化物之间主要靠较弱的分子间结合力吸引,吸附能力较弱,致使正极对多硫化物的捕捉能力较低14,15。对此,研究人员在碳材料上引入各种官能团,加强对多硫化物的吸引,减少活性物质的损失16。除了正极材料,对于锂硫电池其他组成部分的研究也在不断进行中,下面将逐一展开。
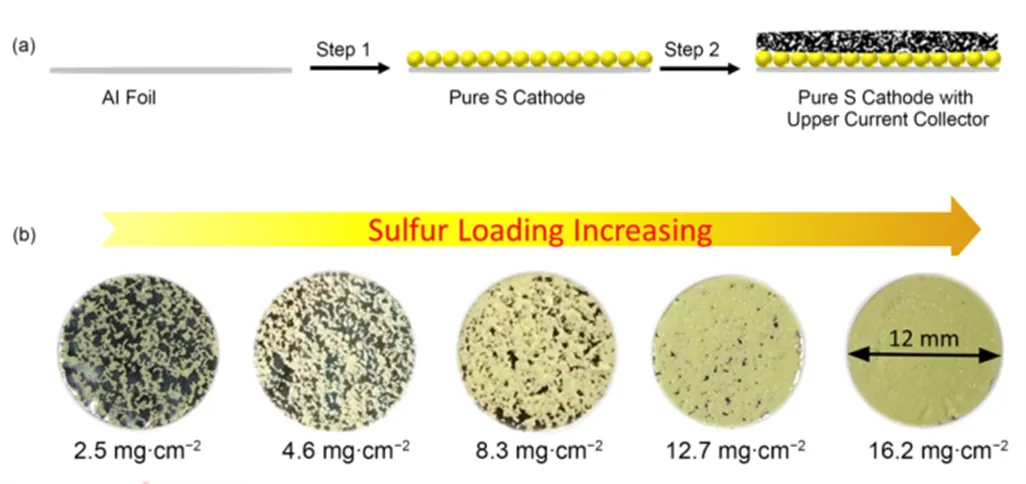
图1 (a)纯硫电极的制造示意图及(b)硫负载量从2.5至16.2 mg∙cm−2变化的纯硫电极图像
3 正极材料
单质硫是电子绝缘体(5 × 10−30S∙cm−1,25 °C),不能单独作为电极材料使用,并且硫的最终放电产物Li2S、Li2S2也是电子绝缘体,一旦生成的Li2S、Li2S2包裹在电极材料表面,会使电子无法传递到材料内部,导致电池性能急剧下降。2016年,Manthiram等17首次使用商业硫磺粉末作为活性材料,采用Blade-Cast方法制备出了高面积负载纯硫电极。制备过程如图1(a)所示。首先,将硫磺粉末(95%,质量分数)和聚偏二氟乙烯(PVDF,5%,质量分数)粘合剂混合,充分分散在-甲基-吡咯烷酮(NMP)中,然后使用Blade-Cast方法将其涂覆在Al箔上,通过限制NMP溶剂的量,可以得到高面积负载纯硫电极,如图1(b)所示。
3.1 硫/碳复合材料
目前,对锂硫电池正极材料改性上,硫/碳复合材料是研究的热点。碳材料具有良好的导电性,还具有高孔隙度、强吸附能力、低成本等优点,利用碳材料组建导电网络,能够弥补硫单质的绝缘缺陷;利用碳材料的多孔性,使硫能够均匀分布到碳材料间隙中,从而提高硫的负载量,同时丰富的空隙也提供了更多的活性位点;碳材料中分布着复杂的孔结构,能够物理限制多硫化物,阻止多硫化物的溶解和扩散,抑制“穿梭效应”;利用碳材料优异的机械强度和多孔结构,在一定程度上能够缓解锂硫电池充放电过程中造成的电极体积膨胀和收缩。目前,研究较多的硫/碳复合材料有硫/多孔碳复合材料、硫/碳纳米管复合材料、硫/石墨烯复合材料、硫/碳纤维复合材料等,下文将对它们进行详细介绍。
3.1.1 硫/碳纳米管复合材料
碳纳米管具有比表面积大、导电性强、微孔结构丰富及机械强度高等优点,是最早用在锂硫电极正极中的碳材料。Lee等18通过研究发现,当硫单质和碳纳米管(20%,质量分数)复合作为锂硫电池的正极时,放电比容量可以提高到485 mAh∙g−1(未复合时放电比容量仅为400 mAh∙g−1)。但是在他们的研究中,硫和碳纳米管并不能很好地紧密接触。
由于155 °C条件下硫的粘度很低,将硫和碳纳米管在155 °C下混合,能够获得很好的贴合度。和室温下简单混合制备成的硫/碳纳米管复合正极相比,155 °C条件下制备的硫/碳纳米管复合正极表现出更低的电荷转移阻抗和更好的循环性能。Qiu等19通过热处理法得到了硫包覆的碳纳米管复合材料(S@CNT),很大程度上提高了硫的利用率和循环稳定性。
之后,研究人员发现杂原子改性碳材料表面的方法能够进一步提高锂硫电池的电化学性能。Zhang等20使用氮掺杂的碳纳米管作为正极载硫材料,得到了稳定循环的锂硫电池,1倍率下循环200次仍有937 mAh∙g−1的放电比容量,容量保持率为70%。Manthiram等21将羟基化的氮掺杂碳纳米管(H-NCNT)用作锂硫电池正极的载硫材料,利用H-NCNT中的聚硅氧烷与氮和羟基之间的相互作用来减弱“穿梭效应”。这种具有2.2 mg∙cm−2硫的碳硫复合电极显示出优异的性能(/5倍率下的初始放电比容量为1341 mAh∙g−1,5倍率下的初始放电比容量为849 mAh∙g−1),500次循环后稳定性仍然十分良好(每次循环衰减0.06%)。Manthiram等22制备出了S-HMT@CNT,结构的示意图如图2。中空TiO2球和碳纳米管进行复合用于载硫,中空TiO2用来吸附硫,碳纳米管用来提高导电性,均匀的复合结构保证材料具有较好的强度,双功能碳纸夹层(DF-PCW)改善了材料的浸润性。
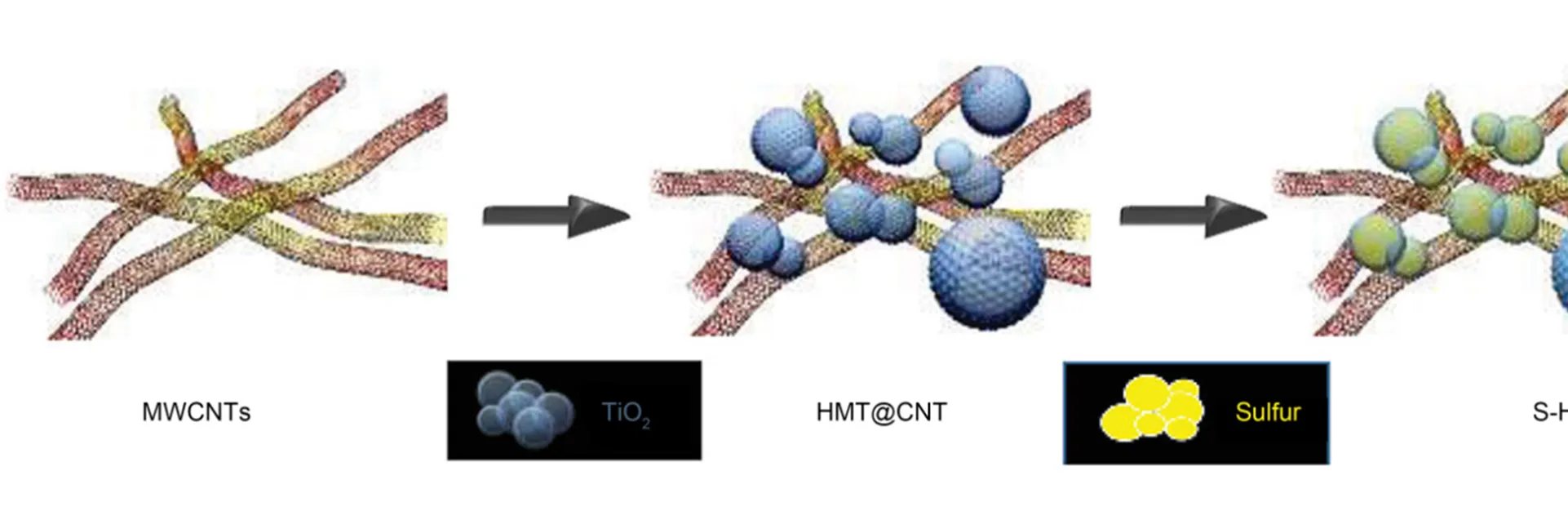
图2 HMT和CNT中硫含量比例的详细示意图
为了提高锂离子的扩散效率和硫的负载率,Lin等23将金属有机骨架和碳纳米管进行交叠,用在没有粘结剂存在的锂硫电池正极中。高度多孔的金属有机骨架不仅提高了正极的载硫能力,内部的导电通道也提高了离子传输效率。这种分层多孔的三维导电网络具有很高的体积能量密度。
3.1.2 硫/石墨烯复合材料
石墨烯是一种由单层碳原子构成的二维材料,具有比表面积高、电导率高、质量轻、结构强度高等优点。石墨烯可以有效包覆硫颗粒,形成导电网络,保证离子的有效传输,从而使界面阻抗降低,大幅度提高正极材料的电化学活性。将石墨烯引入正极材料主要有以下几个途径:石墨烯或者氧化石墨烯直接作为负载硫单质的碳基体;在石墨烯上引入官能团,减少正极活性物质的损失;改变石墨烯的层间结构,优化空间结构,提高正极硫负载率。
Li等24将孔隙率为3.51 cm3∙g−1的高度多孔石墨用作锂硫电池的正极材料,载硫率得到很大提高(80%,质量分数),正极结构如图3所示,其中将具有高导电性的石墨烯作为集电极和活性物质吸附层,电池初始放电比容量高达1500 mAh∙g−1。
Dai等25在Triton X-100聚合物存在的情况下将硫代硫酸钠与盐酸进行反应获得硫颗粒,随后加入石墨烯和碳黑以形成复合材料。其中石墨烯外壳用于捕获锂多硫化物物质并提高电极整体的导电性,而Triton X-100聚合物有助于减少硫的体积膨胀。与不含石墨烯的正极材料相比,石墨烯/硫复合电极提高了锂硫电池的循环稳定性。Zhang等26利用化学沉积法制备了S-氧化石墨烯复合材料,在电化学反应过程中,氧化石墨烯表面的官能团能够吸附硫及多硫化物。Nazar等27通过理论计算,氧化石墨烯表面的羟基可以加强C―C键和S原子的结合力,能够有效减少正极活性物质的损失。Chen等28通过在还原氧化石墨烯上负载超小型硫纳米颗粒,极大的提高了正极载硫量。硫首先与乙二胺反应形成硫胺络合物,然后在盐酸条件下将该络合物加入到还原的氧化石墨烯中,络合物分解到石墨烯片的表面上形成硫纳米颗粒,通过控制pH和反应时间,可以获得直径为5–150 nm的硫纳米颗粒,0.1下得到了1672 mAh∙g−1的初始放电比容量,0.5和1下的长循环分别得到1661和1574 mAh∙g−1的超高放电比容量。
Zhang等29通过对石墨纳米片进行改性,制备成具有纳米孔结构的石墨烯材料。纳米孔的体结构对于硫及中间产物的吸附能力远远大于表面吸附。这种孔结构是良好的电子导体和离子导体,能够束缚体积相对较大的多硫化物,不仅有效减少了硫的损失,而且缓解了充放电循环中硫体积改变造成的破坏。Wei等30制备了非堆叠结构的双层石墨烯,首先,通过化学气相沉积在介孔金属氧化物两侧沉积石墨烯层。然后去除金属氧化物模板,获得非堆叠的双层石墨烯,硫颗粒在介孔以及层中均匀分散。这种非堆叠结构的石墨烯-硫复合材料具有较高的比表面积和良好的导电性,能够促进离子的快速转移。在5和10的高倍率下,分别获得了1034和734 mAh∙g−1的高比容量,1000次循环后均获得了52%容量保持率。
Huang等31用氨溶液加热氧化石墨烯的方式制备出了氮掺杂的石墨烯(NG)。氮掺杂后的石墨烯显示出1.02 S∙cm−1电子电导率,比未掺杂的石墨烯高出一个量级。S@NG复合材料在0.4倍率下循环多次后仍具有稳定的循环性能。Zhang等32设计合成了一种S@NG纳米复合材料,其中超细纳米硫颗粒包裹在高导电性NG片中。将该S@NG纳米复合材料用作锂硫电池的正极,在不加入其它碳添加剂的情况下,5高倍率下的初始放电比容量为606 mAh∙g−1,同时在2倍率下的循环寿命超过2000次。
图3 硫正极全石墨烯结构设计示意图
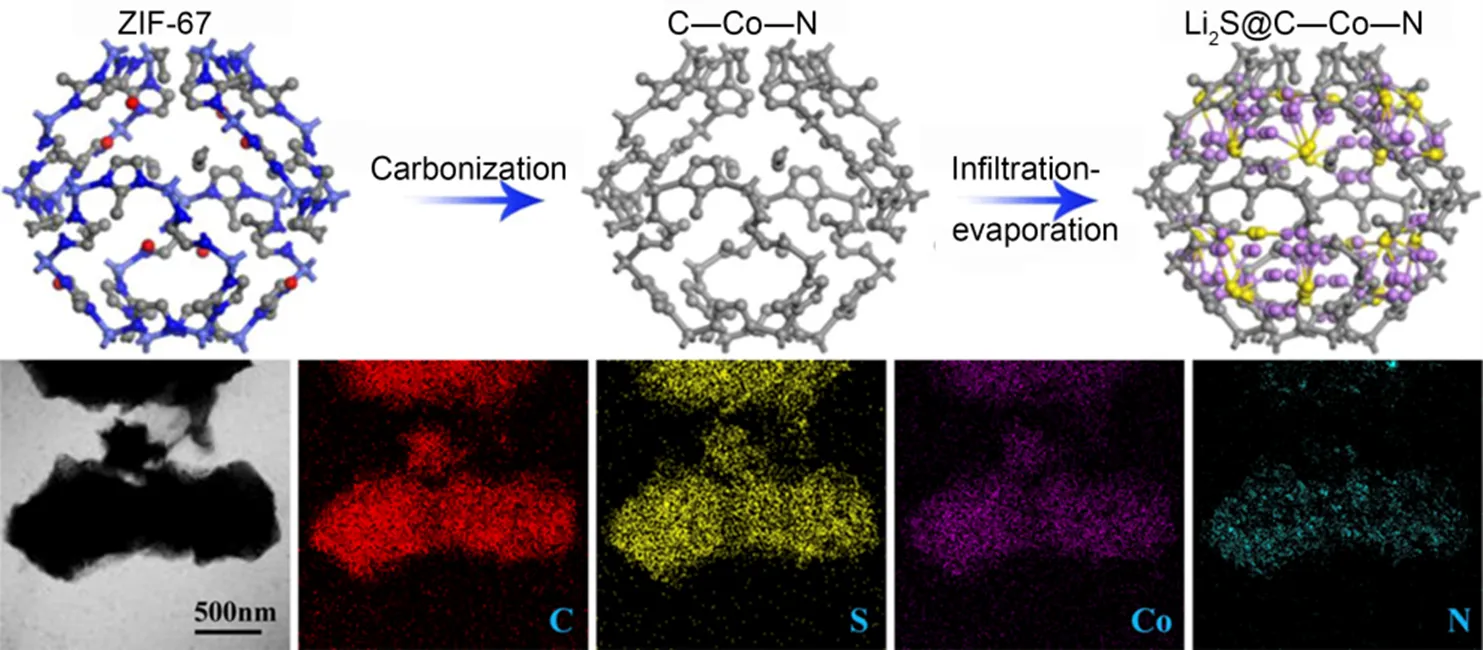
图4 Li2S @ C―Co―N复合材料合成工艺示意图
3.1.3 硫/多孔碳复合材料
根据孔尺寸的大小可以将多孔碳可分为微孔(< 2 nm)、介孔(2–50 nm)和大孔(> 50 nm)三类。多孔碳材料的多孔结构能够物理限制活性材料,同时多孔碳具有较高比表面积,用作正极材料碳基体能明显改善锂硫电池的电化学性能。
Yan等33通过液相沉积法合成了微小的硫颗粒,并和活性多孔碳复合后用作正极材料。与传统载硫方式相比,这种方法工艺简单、操作方便、成本低。得到的电池在/10下具有1093 mAh∙g−1的初始放电比容量。Guo等34用活性碳球作为硫载体,通过调节KOH溶液浓度,能够得到不同孔直径的活性碳球。
Kim等35系统研究了介孔碳对电池性能的影响。介孔碳具有高导电性和高孔隙等优点,用作正极碳基体能够明显提高电池的循环性能,说明介孔碳可以抑制硫的损失和多硫化物的扩散。进一步研究表明,介孔碳的孔径作用不明显,而孔容能够有效提高电池性能。
具有金属离子的金属有机骨架(MOFs)多孔碳材料具有丰富的纳米尺度空穴和开放通道,有利于小分子的进入。He等36将MOF多面体(ZIF-67)进行碳化,获得了由MOF衍生体和钴、氮(C―Co―N)共掺杂的结构体,并最终得到了高导电性石墨碳,合成示意图如图4所示。由于C―Co―N和Li2S纳米颗粒之间具有协同相互作用,Li2S@C―Co―N复合物在初始循环时可以获得1155.3 mAh∙g−1(理论值的99.1%)的超高可逆放电比容量,在300次循环后放电比容量仍为929.6 mAh∙g−1,具有近100%的库仑效率。
3.2 硫/金属化合物复合材料
3.2.1 金属氧化物
含有O2−阴离子的纳米金属氧化物通常具有很强的极性表面,由于氧和金属之间的强结合,金属氧化物不易溶于大多数的有机溶剂。与纳米结构碳材料相比,金属氧化物为多硫化物的吸收提供了丰富的极性活性位点。由于其内在缺陷和独特的带结构,金属氧化物具有更好的导电性。此外,氧化物能够显著增加锂硫电池的体积能量密度。
TiO2是天然存在的钛的氧化物,具有较好的极性表面,能够广泛用在锂硫电池正极中。具有不同形貌的纳米结构TiO2先后得到研究,发现其用作锂硫电池正极能够极大改善电池性能,例如介孔中空TiO2球体、纳米颗粒、纳米管等。Guo等37制备出了载硫的介孔TiO2球,所制备的介孔TiO2球体具有较大的孔体积,这提供了充足的内部空间用于储存硫,同时也为电池反应提供了巨大的反应界面,加快了电子传输。
受TiO2的启发,随后研究人员提出了将Ti4O7用作锂硫电池的正极材料添加剂,并证明了Ti4O7在用于锂硫电池正极材料添加剂时能够有效改善电池性能。Caruso等38通过原位碳热还原法制备出了介孔的Magnéli相Ti4O7微球,它们具有相互连接的介孔(20.4 nm)、较大孔体积(0.39 cm3∙g−1)和较高的比表面积(197.2 m2∙g−1)。图5是原位碳热还原法合成介孔的Magnéli相Ti4O7微球的示意图,当硫阴极嵌入介孔Magnéli相Ti4O7微球的基体中时,中等倍率(/10)下表现出优异的可逆比容量(1317.6 mAh∙g−1),400次循环后仅衰减了12%。
Zhang等39通过简单的机械混合法合成了硫/V2O5复合材料作为锂硫电池的正极材料,这种材料能够促进离子的迁移,降低电极的电化学阻抗。但是受到“穿梭效应”的限制,初始放电比容量相对较低,不能满足实际需要。Kim等40首次证明了具有表面羟基的亲水性氧化镁(MgO)纳米粒子可以用作正极材料添加剂来吸附锂硫电池反应过程中的多硫化物,并证明了MgO纳米颗粒能够均匀分布在活性硫的表面。与纯硫电极相比,向正极中添加MgO使锂硫电池的活性物质容量衰减速率降低,循环稳定性大大提高。这归因于MgO与锂聚合物之间的强化学作用,从而抑制了“穿梭效应”,提高了硫的利用率。
金属氧化物除了能够用作锂硫电池纯硫正极添加剂之外,也可以将其直接添加到碳复材料中去,得到金属氧化物和碳的复合正极材料。Long等41通过将超细Nb2O5纳米晶体复合到介孔碳框架上,在物理和化学层面上有效的减少了活性硫的损失。电化学动力学分析进一步表明,Nb2O5纳米晶体可以作为电催化剂,显著加速多硫化物之间的氧化还原反应,Nb2O5能够将可溶性Li2S6/Li2S4还原成不溶性Li2S2/Li2S,通过克服氧化还原反应的动力学障碍,得到的介孔碳微球/Nb2O5/S复合材料在较低的极化、较高的库仑效率和较高的速率能力方面能够较好地改善电化学性能。0.5下具有1289 mAh∙g−1的高初始比容量,200次循环后可逆比容量为913 mAh∙g−1。更重要的是,高电流密度下的电化学性能是目前最好的,5下具有887 mAh∙g−1的可逆比容量,2条件下500次循环后的可逆比容量为650 mAh∙g−1。

图5 溶解渗透介孔锐钛矿TiO2微球,利用原位碳热还原制备介孔Magnéli相Ti4O7微球的示意图
3.2.2 金属硫化物
随着金属硫化物合成方法的逐渐丰富,研究人员开始尝试将其加入到锂硫电池正极中去。金属硫化物对含硫物质具有较强亲和性,能够很好地限制正极活性物质的损失。
黄铁矿CoS2在多硫化物还原过程中具有较高的催化活性,同时黄铁矿型CoS2晶体在300 K时具有6.7 × 103S∙cm−1的电导率,用于锂硫电池正极材料在理论上是完全可行的。Jin等42用CoS2(15%或30%)和石墨烯(G)材料混合构成了CoS2/G复合正极材料,通过电化学分析证明CoS2能够作为有效电催化剂,加快锂硫电池多硫化物的氧化还原反应进程。同时通过调节CoS2的比例能够改变电池的初始放电比容量,在低硫负载(0.4 mg∙cm−2)下,对于CoS2(30%)/G正极,初始放电比容量为1174 mAh∙g−1;对于CoS2(5%)/G正极,初始放电比容量为1368 mAh∙g−1(比S/G正极高62%)。
Pyun等43制备出了MoS2颗粒状夹杂物和硫的复合正极材料,并证明了MoS2夹杂物能够有效缓解多硫化物的溶解。用这种复合正极材料组装成的锂硫电池,循环寿命达长达1000次,并且具有较低的容量衰减率(每次循环仅衰减0.07%)。Ding等44将NiS2非晶添加到锂硫电池正极中去,得到的锂硫电池在0.1 A∙g−1下表现出高达1540 mAh∙g−1的初始放电比容量,0.5 A∙g−1下循环1200次后仍有77%的容量剩余。
Xiong等45利用一步水热法在碳纳米纤维(CNF)上沉积WS2纳米片,得到了C@WS2的复合材料,然后在155 °C条件下负载硫,最终获得C@WS2/S正极材料,合成步骤如图6。其中CNF为非极性材料,主要负责电子传输,WS2纳米片为极性材料,有较高活性表面和较低的接触电阻,整体的纤维结构保证了锂硫电池具有较好强度和韧性。
黄铁矿FeS2是自然界中硫矿物中含量最高的,用FeS2作合成材料能够大大减少生产成本。Zhang等46已经通过实验证明了黄铁矿FeS2对多硫化物具有优异的化学吸附能力,其中FeS2中的硫位点可以和多硫化物自由基阴离子相互作用形成活性络合物。
在所有的金属硫化物中,对硫化锂正极材料的研究是最为广泛的。传统锂硫电池中,由于硫复合正极不含锂,需要金属锂负极提供锂源,这样锂负极在反应过程会发生结构变化,给锂硫电池体系带来安全隐患。研究人员开发硫化锂材料来代替纯硫正极材料,很好地消除了金属锂负极结构变化给锂硫电池带来的影响。Cai等47运用高能干燥球磨法制备了纳米级Li2S/C复合正极材料,该电池的初始放电比容量为1144 mAh∙g−1。
其它硫化物同样可以用作锂硫电池的正极材料,例如MnS48,CuS49,ZnS50,SnS251,NiS252等。但是硫化物普遍导电性差,必须同其他导电性好的材料复合。
3.2.3 其他金属化合物
大多数金属氧化物导电性差,仅使用金属氧化物作为锂硫电池正极材料可能会阻碍电子传输,导致硫利用率和倍率能力降低。与此同时,氮化钛(TiN)由于其高导电性和优异的化学稳定性引起了研究人员的注意。Goodenough等53通过固相分离法合成了具有较高比表面积的介孔TiN,并将其用于锂硫电池正极中。TiN-S复合正极具有高导电性、坚固的多孔骨架和较好的吸附能力,组装成的锂硫电池显示出极佳的循环稳定性,500次充电/放电循环中,单循环容量衰减仅为0.07%。Huang等54同样使用TiN作为锂硫电池正极材料的添加剂,0.5倍率下可逆比容量为1012 mAh∙g−1,即使在5倍率下,可逆比容量仍有550 mAh∙g−1。

图6 在CNF上垂直对齐的WS2的制备示意图
最新研究表明金属碳化物对多硫化物具有很强的吸附作用,如已经研究过的MXene相Ti2C55、TiC56。Arava等57的研究证明纳米结构过渡金属碳化物(TMC),如TiC和WC可以吸附聚合物,从而促进多硫化物还原反应的发生。TiC正极材料由于具有微晶尺寸(约25 nm)和大量活性位点,用在锂硫电池正极中能够提高循环性能,100次充放电循环中具有860 mAh∙g−1的稳定比容量,这对下一步的研究具有指导意义。
3.3 硫/导电聚合物
导电聚合物在锂硫电池系统中也得到了广泛应用,导电聚合物自身有很多优点:优良的导电性,能够促进电子传输效率;较好的韧性,在循环期间可以缓解硫的体积变化。
在最开始对导电聚合物的研究上,研究人员大多把聚吡咯(PPY)作为锂硫电池正极的涂层材料。Wang等58发现PPY的存在能够提高锂硫电池正极的导电性,同未加PPY的正极材料相比,正极为S-PPY复合物的锂硫电池得到了更高的放电比容量。但是随后的20次循环的容量衰减速率并没有得到改善,这表明仍然有多硫化物溶解到电解质中。
为了提高正极材料对多硫化物的吸附率,研究人员59在硫颗粒表面上涂覆均匀聚合物,制备成了硫-聚噻吩(PT)的核-壳复合材料。通过控制加入的噻吩单体的量,制备成具有38.0%、28.1%和17.5% PT的3种不同复合物。在电化学性能研究上,与未涂覆的情况相比,PT涂覆过正极的锂硫电池显示出更好的循环稳定性,0.06倍率下进行80次循环后,仍具有74%容量保持率,证明了均匀PT壳能够有效限制多硫化物的溶解。
为了进一步提高锂硫电池的循环性能,Liu等60将聚苯胺(PANI)纳米管应用于正极材料。在渗透硫的过程中,硫与PANI反应形成三维交联网络,其具有链间或链内二硫键结构,这种结构有助于在分子水平上将硫固定。由于这种独特的限制效应,S-PANI纳米管的比容量可达568 mAh∙g−1,在1的电流密度下循环500次后仍具有76%的容量保持率。反应结束后正极材料的体积没有发生明显改变,这表明柔性聚合物结构能够缓解硫在循环过程中的体积变化。
Abruna等61为了进一步减小硫化物在锂化过程中的体积膨胀问题,合成了具有内部空隙空间的硫/聚苯胺(S/PANI)蛋黄纳米结构。通过对硫/ /PANI进行热处理,硫与PANI壳反应形成了三维交联的二硫化物网络,从而使硫/聚苯胺的“核壳”结构转变成“蛋黄”结构。“蛋黄”结构中具有大量的内部空隙,这些空隙缓解了正极材料在电池循环过程中的体积变化,继而提高了锂硫电池的电化学性能。在0.2和0.5倍率下放电比容量分别达到了1101和920 mAh∙g−1,且在200次循环后仍具有70%和68%的容量保持率。
除了使用蛋黄结构形态,制备具有中空结构的正极材料也可以缓解硫的体积变化。Cui等62将硫代硫酸钠与盐酸在聚乙烯吡咯烷酮(PVP)中进行反应。PVP将组装成中空球形囊泡胶束,其中疏水性碳主链指向内部,亲水性酰胺基团指向外部。由于硫本质上是疏水性的,它将在胶束的内部疏水部分上优先成核和生长,导致中空硫纳米颗粒的形成。然后分别将三种不同的导电聚合物PEDOT,PANI和PPY涂覆在纳米颗粒上。结果表明用PEDOT涂覆的正极材料组装成的电池性能最好,在0.5下循环500次后,比容量高达1165 mAh∙g−1,容量保持率为67%。
结构上的改变对性能的提升是有限的,为了进一步减少多硫化物的损失,可以在结构优化的基础上向导电聚合物正极增加官能团。Su等63首先将MnO2纳米片官能化,然后将其引入到导电聚合物核-壳纳米球中制备成了锂硫电池的正极材料(S@PEDOT/MnO2),合成方案如图7所示。其中PEDOT层提高了导电性,PEDOT上的MnO2纳米片提供提高了活性接触面积,增强了电解质与电极材料的润湿性。结果显示,与S@PEDOT (551 mAh∙g−1)相比,S@PEDOT/MnO2在0.2下200次循环后的比容量为827 mAh∙g−1,性能提高了50%。
4 电解质
电解质作为锂硫电池中重要的组成部分,在离子的传递上起着十分重要的作用。另外,多硫化物的“穿梭效应”也是发生在电解质中的,这对接下来的研究很有启示,研究新型的电解质,在保证较高锂离子传导性的前提下,避免充放电过程中形成的多硫化物(Li2S6、Li2S4)的“穿梭效应”,提高活性物质的利用率,从而提高锂硫电池的循环性能。
近几年的文献报道大多是关于正极材料的结构改性64,65,研究成果当然是不可否认的,但是鉴于电解质在锂硫电池离子迁移中的关键作用,研发新的电解质来提高锂硫电池的性能也是很好的研究方向。锂硫电池的理想电解质应具备诸如较高的离子传导能力、较好的电子绝缘性、较宽的电化学窗口、稳定的化学性能以及和正负极稳定的化学反应活性等性质。新型电解质材料的种类繁多,机理相异,下面将详细介绍其进展。
4.1 液态电解质
液态电解质是最早进行研究的电解质类型,一般液态电解质的成分都包含有溶剂、溶质、溶解盐以及增强其性能的添加剂等。传统电解质的研究主要集中在碳酸盐、砜等溶剂上,主要将乙二醇二甲醚(DME)66、四乙醇二甲醚(TG)67和环状醚类溶剂DOL68、甲基乙基砜(EMS)等溶剂按一定比例混合。但是仅靠传统电解质溶剂并不能解决多硫化物的穿梭问题,这种电解质的溶剂甚至会和多硫化物发生反应,造成硫的巨大损失,所以有必要开发出新型的溶剂来代替这些传统的溶剂。常见的方法就是使用极性较弱的溶剂,这样有利于减少对多硫化物的吸引和溶解。Zhang等69使用有机氟复合物1,1,2,2-四氟乙基-2,2,3,3-四氟丙基醚(TTE)作为溶剂,与传统电解质相比,使用这种电解质的锂硫电池电化学性能得到提升,作者将其归因于TTE较弱的极性。Lai等70将1,1,2,2-四氟乙基乙醚作为电解质,在电池放电过程中,这种电解质会在锂电极表面产生一层保护膜,这种保护膜阻止多硫化物沉积到锂电极上,使多硫化物的溶解度降低,并且这种电解质不和多硫化物进行反应,这样活性物质的质量得以提高。
此外,在LiTDI基电解质中添加0.1%的Li2S和0.2 mol∙L−1的LiNO3,能够提高锂硫电池正极硫的负载率。Belharouak等71将0.2 mol∙L−1Li2S和0.5 mol∙L−1LiNO3加入到DME溶剂中,证明了锂盐添加物能够有效抑制锂硫电池的“穿梭效应”。Aurbach等72发现,在电解质中加入低浓度的聚硫化合物添加剂可以在锂负极产生一层Li2S保护膜,阻隔锂负极和溶液中多硫化物的进一步接触。添加剂的主要作用是对负极的保护,本文的负极部分将会详细阐述。
4.2 固态电解质
固态电解质能够抑制“穿梭效应”,同时提高锂硫电池的安全性,从而引起了广泛关注。但是到目前为止,较低的离子电导率仍然是固态电解质应用的主要障碍。
4.2.1 聚合物电解质
聚合物电解质具有以下优点:(1)和电极接触更加紧密;(2)化学和电化学稳定性更好;(3)能够在分子水平上进行结构设计。聚合物电解质通常可分为全固态聚合物电解质(SPE)和凝胶聚合物电解质(GPE),这里只讨论SPE类型的电解质。
SPE由锂盐和高分子量聚合物基体组成,简言之,就是锂盐直接溶解于聚合物基体中而形成的固态体系。全固态聚合物电解质离子电导率低,这不能满足锂硫电池实际应用的需求。
Wen等73将含(PEO)20LiTFSI-10% ()-LiAlO2型SPE应用于锂硫电池。对电极进行放电测试,0.1 mA∙cm−2电流密度条件下初始放电比容量为450 mAh∙g−1。随后该研究小组74将(PEO)18LiTFSI-10% ()SiO2型SPE用于正极材料为硫/介孔碳球的锂硫电池中,首次放电比容量为1265 mAh∙g−1。
在室温下,PEO的高结晶度大大降低了离子电导率。而半结晶聚合物电解质的导电性主要通过络合物中的无定形相实现,这就限制了PEO的实际应用。对此研究人员已进行了各种尝试,包括合成具有较低结晶度和较低玻璃化转变温度的新聚合物,以获得高电导率的SPE。Balsara等75研究了SEO和锂聚合物混合物的相行为。聚合物的添加会导致SEO从无序到有序以及有序到无序的相变,形态变化取决于盐浓度、阴离子大小以及聚合物的质量分数。这项研究说明嵌段共聚物电解质具有应用于锂硫电池的潜在可能性。
若能在可接受温度范围内提高SPE电解质的电导率,聚合物电解质将在锂硫电池甚至传统的锂离子电池的应用具有很大的竞争力。
4.2.2 无机固态电解质
在锂硫电池中,无机固态电解质能形成物理隔层保护锂负极,能够阻止S2−(4 ≤≤ 8)向金属锂电极的扩散。并且,在很大的温度范围内无机固态电解质都可以保持很好的化学稳定性。
Hayashi等76在S/CuS作为正极、Li-In合金作为负极的情况下使用Li2S-P2S5玻璃陶瓷电解质作为电解质,组装的全固态锂硫电池在0.064 mA∙cm−2放电电流密度下经20次循环后的放电比容量为650 mAh∙g−1。
Machida等77用高能球磨法制备60Li2S- 40SiS2玻璃电解质,将其与Cu/S复合材料正极、Li4.4Ge负极组装成锂硫电池,在0.64 mA∙cm−2放电电流密度下的首次放电比容量大于980 mAh∙g−1。之后Hassoun等78采用同样的方法制成Li2S-P2S5玻璃电解质,以硫/石墨烯为正极,金属锂为负极组装成锂硫电池,在/20倍率下进行放电测试,发现20次循环中放电比容量始终保持在400 mAh∙g−1左右,能量密度为840 Wh∙kg−1。Tu等79通过高能球磨加退火的方法制备了一种新型MoS2掺杂的Li2S-P2S5玻璃-陶瓷电解质(Li7P2.9S10.85Mo0.01)。室温条件下,Li7P2.9S10.85Mo0.01具有4.8 S∙cm−1的高离子电导率,与原始的Li7P3S11电解质相比,Li7P2.9S10.85Mo0.01使锂金属在电池循环过程中的稳定性更好。电池循环测试显示,使用这种电解质的锂硫电池具有高达1020 mAh∙g−1的放电比容量,远远优于Li7P3S11电解质。
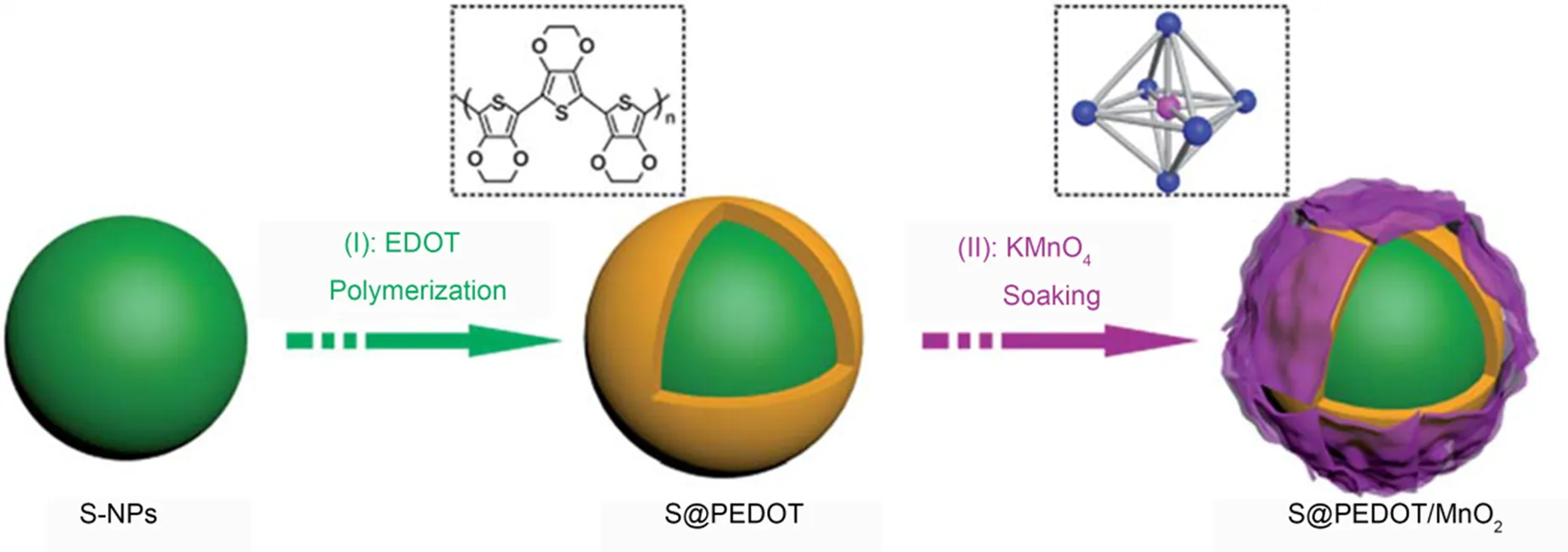
图7 S@PEDOT/MnO2复合材料的合成示意图
Chen等80将凝胶陶瓷多层电解质用于锂硫电池的隔板和电解质,发现较弱的“穿梭效应”,电池表现出良好的电化学性能,几乎没有自放电行为。电池表现出高达725 mAh∙g−1的初始放电比容量,/2下300次循环后放电比容量仍保持在700 mAh∙g−1。
无机固态电解质的锂离子迁移数接近于1,理论上能完全阻隔S2−(4 ≤≤ 8)向金属锂负极的扩散。但是无机固态电解质制备过程复杂、机械强度低、与电极接触的界面阻抗较大等缺点限制了无机固态电解质的实际应用。
4.2.3 离子液体电解质
离子液体电解质完全由离子组成, 具有锂离子电导率高、电化学稳定性好、溶解性好等优点。并且与上述电解质相比,离子液体电解质不易挥发和燃烧,能够显著提高电池的安全性。
Zhang等26利用化学沉积法制备了硫-氧化石墨烯复合材料(GO-S),将其用于锂硫电极正极中,组装的电池具有优异的容量稳定性。同时,Zhang等人使用了离子液体电解质,使该电池的倍率性能达到了2。
Wan等81应用室温离子液体-甲基--丙基吡咯烷鎓-双(三氟甲烷磺酰)亚胺(RTIL P1A3TFSI)作为电池的电解质。该电解液具有不可燃性、蒸气压低、锂离子电导率高、电化学窗口宽等优点,这使得组装成的电池化学性能十分稳定,在0.1下的放电比容量高达1457 mAh∙g−1。受此启发,Xia等82发现-甲基--丙基哌啶鎓双(三氟甲磺酰基)亚胺(PP13TFSI)电解质对多硫化物的穿梭有明显的抑制作用。这种电解质可以使充满电的锂硫电池在两天内实现零自放电。如图8所示,大多数Li2S8溶解在DOL/DME溶剂中,沉淀可忽略不计,产生深棕色溶液。在PP13TFSI/DOL/DME的溶剂中,在小瓶底部观察到更多的沉淀,说明PP13TFSI能够减少多硫化物的溶解。
Watanabe等83制备了两种离子液体电解质:双三氟甲磺酰基酰基锂/[,-二乙基--甲基--(2-甲氧基乙基铵)]与双三氟甲磺基胺Li[TFSA]/[DEME][TFSA]混合液离子液体电解质和双三氟甲基磺酰基氨基锂/四甘醇二甲醚Li[TFSA]/(TEGDME)离子液体电解质。Li2S在0.98 mol∙L−1Li[TFSA] /TEGDME溶液中的溶解度高于Li[TFSA]/[DEME][TFSA],这表明这种离子液体电解质对“穿梭效应”的抑制效果优于Li[TFSA]/TEGDME溶液,这归因于Li[TFSA]/ [DEME][TFSA]溶液中的[TFSA]−阴离子具有较弱的路易斯碱性,可以使Li2S有非常低的溶解度。Chen等84设计了含有-甲氧基乙基--甲基吡咯烷鎓-双(三氟甲磺酰)-酰亚胺(Pyr1,201TFSI)和三乙二醇二甲醚(TEGDME)的新型离子液体电解质,并将其和LiTFSI/草酸二氟硼酸锂(LiODFB)二元锂盐混合作为电解质。Pyr1,201TFSI/TEGDME电解质的醚官能化离子中的高度柔性的烷氧基链为邻近分子提供了便利的传输通道85,这提高了Li2S在离子液体中的传输效率。而LiODFB的SEI形成层也可以保护Li负极形成锂电极并减轻穿梭现象,电池的初始放电比容量为1264.4 mAh∙g−1,50次循环后容量仍为911.4 mAh∙g−1,库仑效率超过95%。Byon等86设计了新型的室温离子液体(RTIL),它是由有机电解质和1-丁基-1-甲基-吡咯烷鎓(C4mpyr)阳离子有机溶剂离子液体组成的混合物。其中有机电解质提高了多硫化物的溶解度和离子交换速率,C4mpyr阳离子有机溶剂促使负极表面形成SEI膜,两种溶剂的协调作用极大的提高了锂硫电池的循环性能。Zhou等87将1,1,2,2-四氟乙基2,2,2-三氟乙基醚(TFTFE)加入到甘醇二锂盐为基体的离子液体电解质中(SIL)中,发现氟化醚能够明显改善锂硫电池的循环性能,进一步研究表明,TFTFE在SIL电池系统中的关键作用是增强离子传导能力。
离子液体电解质中加入LiNO3有助于保护锂负极,同时LiNO3能够和多硫化物相互作用阻止多硫化物的溶解。其他锂盐,如双乙二酸硼酸锂(LiBOB),二氟草酸硼酸锂(LiODFB)和氟化锂(LiF)与LiNO3的效果相类似,都能够对锂负极起到保护作用,这将在负极保护部分详细介绍。

图8 (a)电解质溶剂中的多硫化物溶解度试验。 (b)在醚(左)或PP13TFSI(右)溶剂中多硫化物溶解机理
5 多功能隔膜系统
在锂离子电池中,隔膜主要是用来防止正极和负极的直接接触,避免电池发生短路现象。隔膜中的孔道主要负责传递离子,保证电池反应的循环进行。此外,通过对隔膜进行多功能设计,还可以抑制多硫化物“穿梭效应”,从而提高锂硫电池的电化学性能。对于隔膜的多功能化设计主要有以下两个途径:对隔膜成分和结构进行改性,提高其离子选择性,从而抑制多硫化物的扩散,阻止穿梭效应的发生;分别对正负极两侧隔膜界面进行改性,从而控制正负极结构,提高稳定性。传统的隔膜主要是烯烃类的隔膜,如聚丙烯微孔膜、聚乙烯微孔膜等。这种隔膜制造简单、强度高,但是无法在高电流密度的情况下保持稳定的充放电性能,而且其电子电导率低,不能阻止多硫化物的扩散,因此有必要研发新型的多功能隔膜系统。
5.1 抑制多硫化物扩散隔膜
为阻止多硫化物的扩散,一些研究者对传统隔膜表面进行改性,主要是在烯烃类隔膜上添加其他物质,提高隔膜的离子选择透过性。Gu等88使用分层组装的方法将PP隔膜层添加在聚烯丙胺盐酸盐(多环芳烃)和(丙烯酸)(PAA)中间。通过控制叠加层数和外部PH值,这种隔膜可以达到很高的离子选择性。电化学测试表明,这种隔膜组装的锂硫电池50次循环后的库伦效率几乎达到100%,初始可逆循环比容量为1418 mAh∙g−1。Manthiram等89在聚合物隔膜表面涂覆了一层轻质碳,在纯硫正极的情况下进行充放电。测试发现:首次放电容量高达1389 mAh∙g−1,200次循环后容量仍保持在828 mAh∙g−1。同时这种锂硫电池具有较低自放电性:放置3个月的电池也只有轻微的自放电现象。
除了对传统烯烃类隔膜表面进行改性外,对新型隔膜的研究也没有停下脚步。由于多硫化物阴离子与锂离子在动力学直径方面上存在差异,通过合理设计隔膜中孔道的大小理论上可以实现离子的选择性通过。Zhou87等提出了以金属有机骨架化合物(MOF)为基元材料的氧化石墨烯复合功能隔膜,采用Cu3(BTC)2型 MOF(HKUST-1)作为“离子筛”,其孔道直径远小于多硫化物的离子直径。氧化石墨烯材料的层间距小于多硫化物的离子直径,因此可以实现锂离子的选择透过性。与纯氧化石墨烯隔膜相比,MOF/氧化石墨烯复合隔膜使锂硫电池的倍率性能得到极大提高。
Wen等90对传统隔膜分别用聚吡咯(PPy)纳米管、PPy纳米线和还原型氧化石墨烯(rGO)进行改性处理,这些导电材料可以抑制电解液中多硫化物的迁移,并降低硫正极的极化,在充放电过程中抑制“穿梭效应”,提高锂硫电池电化学性能,图9是这种电池的隔膜的作用机理。此外,与rGO相比,PPy对锂聚合物的吸附效果更强,PPy改性隔膜对电解液的润湿性更好。使用PPy纳米管改性隔膜的锂硫电池0.5速率下显示出1110.4 mAh∙g−1的初始放电比容量,300次循环后的放电比容量保持为801.6 mAh∙g−1。
对多硫化物具有吸附作用的多功能隔膜也可以实现锂离子的选择透过性。Manthiram等89将活化的碳纳米纤维作为功能层制备出了多功能隔膜。活化碳纳米纤维的微孔结构对多硫化物具有物理吸附作用,能够阻止多硫化物的扩散,抑制锂硫电池的“穿梭效应”。Gerbaldi等91对纤维素进行热聚合得到了具有吸附多硫化物功能的隔膜,提高了锂硫电池对活性物质的利用率。在隔膜中掺杂氮元素也可以提高隔膜对多硫化物的吸附作用。Manthiram等92制备出了聚苯胺纳米纤维和碳纳米管的复合隔膜,这种隔膜对锂硫电池的性能有多方面的提升:多壁碳纳米管的孔道和缺陷能够吸附多硫化物;聚苯胺纳米纤维中的亚胺基团对多硫化物有化学吸附作用;该隔膜还可以作为导电层降低正极表面的反应阻抗。研究人员93–96以聚吡咯纳米线为前体,通过碳化过程获得了氮含量很高(7.12%,质量分数)的多孔碳纳米纤维隔膜。氮原子提高了隔膜的导电性以及正极材料对多硫化物的物理和化学吸附。Wang等97设计了功能性磺化乙炔黑(AB-SO3)涂层隔膜,提高了锂硫电池的整体性能。AB-SO3涂层隔膜在不影响离子传导性的同时,对锂离子和多硫化物具有高度的选择性。
5.2 改善正极侧界面的隔膜
锂硫电池的循环过程中,正极的表面结构会不可避免地发生改变,造成活性材料的损失。在正极和隔膜之间添加碳纸等中间层材料,能够在提高导电性的同时抑制多硫化物的穿梭98,这就是所谓的对正极侧的隔膜进行表面改性,这种改性处理可以降低电阻以及减少活性材料的损失,从而提高锂硫电池的性能。

图9 锂硫电池分离器设计示意图(a)使用普通分离器的电池和(b)带有分离器的电池
Li等99使用导电碳黑涂覆隔膜组装的锂硫电池,在不添加硝酸锂添加剂的情况下,库伦效率可以达到90%以上。电化学阻抗谱显示该多功能隔膜对正极界面的等效电荷传递阻抗(ct)值由未改性前的16 Ω降低至8 Ω。Zhang等100将导电碳层复合到高孔隙率的玻璃纤维隔膜上,大大降低了正极界面阻抗,提升正极的硫利用率,一定程度阻止了多硫化物的扩散。
Zhao等101将氧化多壁碳纳米管(o-MWCNT)涂层)引入到隔膜中,用于改善正极的电化学性能,结构如图10所示。由于多层结构的物理和化学捕获,涂层能够促进电子传导和离子快速迁移,组装的锂硫电池在电流密度为100 mAh∙g−1时拥有高达925 mAh∙g−1放电容量。
充放电过程中正极材料结构会发生变化,多硫化物不可避免地会从正极材料中脱出并扩散至隔膜,最后在正极侧隔膜表面沉积,阻塞正极材料离子传输孔道,降低锂硫电池的电化学效率。Cui等102提出了一种利用导电碳层抑制活性硫材料损失,惰性回收硫组分的策略。这种隔膜在锂硫电池充放电循环后的颜色较浅,表明隔膜中的含硫成分降低。Manthiram等103在隔膜上涂覆微孔碳/聚乙二醇复合材料,这种材料具有丰富的孔结构,能够和电解液紧密接触。并且微孔碳材料对多硫化物具有物理吸附作用,聚乙二醇材料对多硫化物具有化学吸附功能,这种双重吸附作用能够有效减少硫损失。该多功能隔膜的放电比容量为1307 mAh∙g−1,500次循环的单次衰减率低于0.11%。
5.3 改善负极侧界面的隔膜
金属锂负极的枝晶生长问题严重阻碍了锂硫电池的商业化使用,改善锂硫电池锂负极的电化学稳定性一直是研究人员关注的重点,通过对负极侧隔膜进行表面改性处理,可以抑制多硫化物沉积在负极层,避免枝晶生长。
Choi等104用聚多巴胺修饰负极侧隔膜表面,提高了隔膜表面和金属锂负极的亲和性,提高了隔膜的润湿性。聚多巴胺能够调节金属锂负极表面的锂离子浓度场,抑制锂硫电池负极枝晶的生长。Zhang等105用带有极性基团的玻璃纤维修饰隔膜表层,实现了负极锂离子的均匀分布,成功抑制了负极枝晶的快速生长。
通过对负极侧隔膜表面进行改性处理,能够控制锂离子浓度场的均匀分布,有效阻止锂负极的枝晶生长,获得更为安全的锂金属负极。
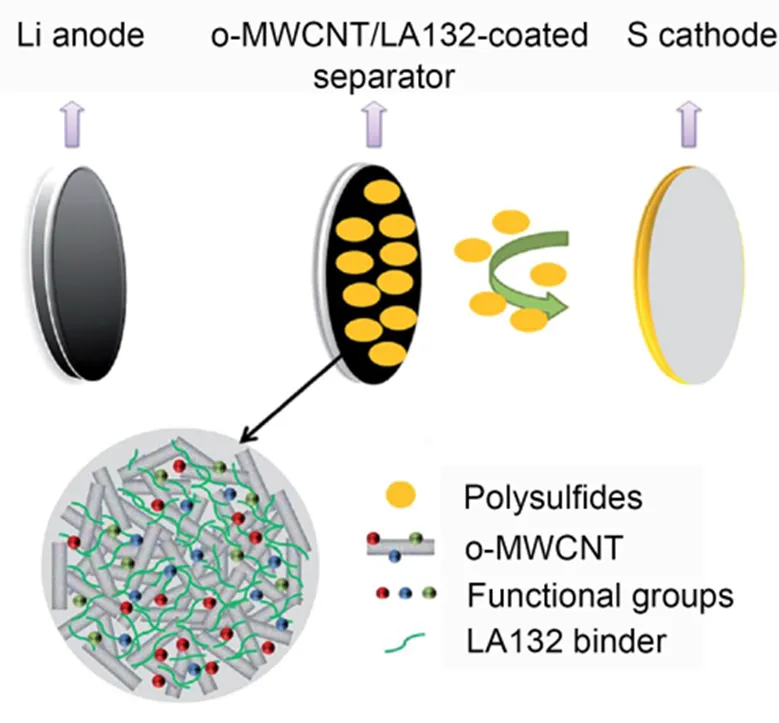
图10 具有o-MWCNT涂层分离器的锂硫电池配置示意图
6 负极保护
金属锂负极的理论容量约为3860 mAh∙g−1,能够极大提高电池的能量密度,常常用作电池的负极材料。但是金属锂性质非常活泼,在锂硫电池循环过程中,容易溶解沉积到电解质中,金属锂的溶解沉积会使电池容量下降,影响电池稳定性。同时随着金属锂的溶解,负极表面会形成锂枝晶,导致粗糙度上升。Chen等106发现最高硫负载量达到99.5%时,锂硫电池容易发生严重的腐蚀现象,造成容量的快速衰减,因此,对锂负极进行保护是十分必要的。目前,负极保护主要主要包括在电解质中加入添加剂、在金属锂表面涂层以及对锂的复合物材料进行替代等方式。下面将一一进行展开。
6.1 电解质添加剂
由于电解质和锂负极是直接接触的,这导致锂硫电池负极侧极其容易发生副反应。研究者在锂硫电池的电解质中加入一些添加剂,希望改善电解质和锂负极的接触环境,在促使锂负极表面形成致密钝化膜,提高电池的电化学性能。
有些添加剂能够降低锂硫电池的自放电频率,避免活性物质快速损失。Liu等107通过研究证实在电解液中添加LiNO3能够减少锂硫电池自放电现象的发生。但是由于硝酸根具有强氧化性,可能降低电池的安全系数,所以研究人员正在努力寻找更为合适的添加剂。
为了防止电解质对锂负极的腐蚀,Wen等108用六甲基二锡对锂负极进行表面改性处理,在负极表面形成了钝化层(SEI层)。改性的锂电极在电解质中表现出更高的稳定性、更低的界面电阻和更低的电荷转移阻抗,电池显示出较好的循环性能,稳定时的放电比容量约为800 mAh∙g−1。
Gao等109将硝酸镧添加剂加入到锂硫电池电解质中。如图11所示,将La(NO3)3引入电解质后,La3+可以立即被Li还原成金属La,继而与聚硅氧烷反应形成La2S3。随着Li2S2/Li2S的沉积,La2S3能够在锂负极形成复合钝化膜,这有利于减少锂负极的电化学溶解/沉积的频率,稳定锂硫电池金属锂负极的表面形貌。显然,硝酸镧添加剂能够有效提高锂硫电池的循环稳定性。
使用电解质添加剂原位形成钝化SEI层能够避免多硫化物与锂金属表面的直接接触,最终提高锂硫电池性能。
6.2 涂层法
在锂负极表面加上涂层材料是最常见的保护负极的方法,为保证涂层材料对锂负极起到有效保护作用,要求涂层材料有良好的化学稳定性、不溶于电解质、不和锂负极及多硫化物发生反应,同时要有良好的离子电导率。
Wu等110在锂负极表面加上凝胶聚合物电解质以及陶瓷膜,陶瓷膜是Li2O-A12O3-SiO2-P2O5- TiO2-GeO2固体膜,这种陶瓷膜阻止了锂负极和电解质的直接接触,电池循环性能得到极大提高。
锂负极在循环过程中出现的锂枝晶制约了电池的进一步循环,严重降低了电池的循环寿命,对负极进行涂层保护能够改善这种状况。Wen等111在负极表面制备导电聚合物层作为保护层,通过导电聚合物层的保护,在醚基电解质和Li负极之间形成稳定且电阻较小的SEI。所获得的电池不仅有效地抑制了锂负极和多硫化锂之间的腐蚀,而且抑制了锂枝晶的生长。在0.5下经过300次循环后,电池的放电比容量保持在815 mAh∙g−1,平均库仑效率为91.3%。
Wu等112制备了由Nafions和聚偏二氟乙烯(PVDF)组成的聚合物,并成功地在锂负极上涂覆这种可以同时抑制Li枝晶生长和多硫化物穿梭的表面涂层,工艺流程如图12所示。这种分级纳米结构复合涂层提供了可靠的机械强度以适应充电-放电循环中Li负极的大体积变化。Nafions/PVDF涂覆的锂负极显著提高了电池循环性能。
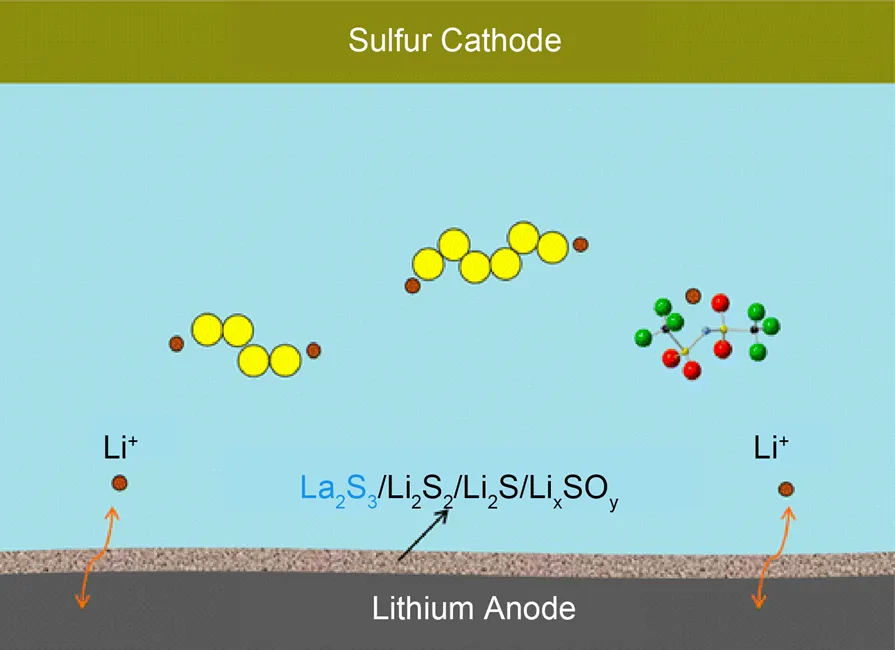
图11 通过向电解液中加入La(NO3)3,Li负极表面形成复合薄膜示意图
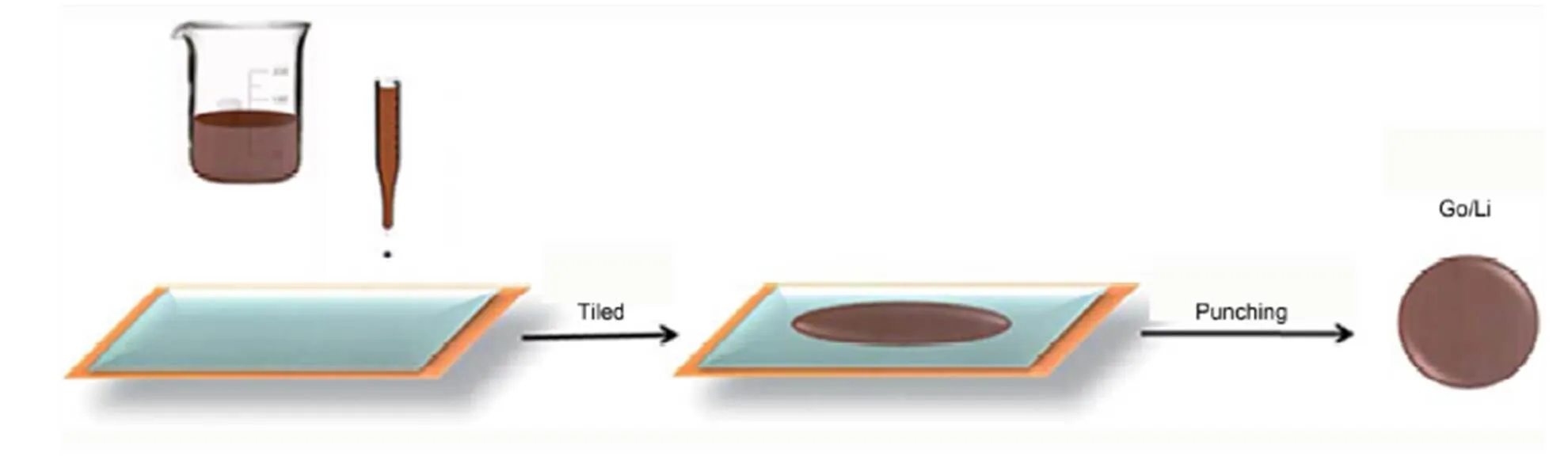
图12 自动铺展方法制作GO/Li电极的工艺流程
Tu等113采用自动铺展方法(工艺流程如图11所示)在金属锂箔上涂覆氧化石墨烯(GO)层,形成复合电极。GO层分布均匀,在金属锂箔上具有较低的粗糙度。通过对GO改性Li(GO/Li)电极的电化学性能进行研究发现,与未改性的纯锂电极相比,GO/Li电极组装的锂硫电池循环稳定性更好。此外,在GO/Li电极中,树枝状锂金属的生长得到有效控制。
涂覆保护层的同时也要考虑保护层对锂离子电导率的影响,保护层应该具有不妨碍离子迁移的性能。Kaskel等114将三元硅碳锂复合材料作为锂硫电池负极,在电池放电过程中,Si/C微球负极能够保存锂离子,并将锂离子输送到硫/碳(S/C)正极。组装的锂硫电池经过1000次循环,比容量仍可达到1470 mAh∙g−1。
负极表面的保护膜可以阻止电解质与锂负极直接发生反应,但在循环过程中保护层持续的稳定性和有效性仍需要进一步研究。
6.3 其他保护方法
通过用锂合金代替锂单质能够防止锂和多硫化物直接接触,从而防止锂的损失。电池循环过程中,负极表面能够形成钝化层,阻止多硫化物和金属锂的直接接触,从而避免负极材料的损失。Wang等115使用Li-B合金作为负极材料,在负极形成了钝化膜,有效降低了由于合金体积变化而造成的性能损失,Li-B合金组装成的电池,性能十分稳定。Scorsati等116用Sn作为负极来代替锂,电池35次循环的可逆比容量为850 mAh∙g−1。Liu等117将石墨引入负极材料中,制成了锂化石墨,锂化石墨和钝化膜作用类似,有利于防止负极副反应的发生,同时锂离子能够在石墨中自由嵌入脱出。
和上述形成保护层机理不同,Wen等118使用氮气直接与锂金属反应形成均匀的氮化锂层(Li3N),形成的保护层(200–300 nm厚)不仅可以有效地抵抗多硫化物的溶解,而且提高了锂离子的传输速率。当被保护的锂负极用于锂硫电池时,0.2下库仑效率高达91.4%,远高于无钝化层负极(库仑效率为80.7%)。
7 前景和展望
7.1 正极材料
正极材料方面,首先为了提高正极的电子和离子电导率,通常是使用导电材料和硫聚合形成复合物,同时为了抑制多硫化物的迁移,在复合材料上引入特殊的官能团,对多硫化物进行吸附。目前常见的正极材料主要硫/碳复合材料、硫/金属化合物、硫/导电聚合物以及上述的一种或几种的组合。
其中在硫/碳复合材料中碳主要形式包括多孔碳、碳纳米管、石墨烯、碳纤维等。对于这些硫/碳复合材料,通过改变结构在一定程度上可以提高正极材料的比表面积,提高硫的负载量,能够提供更多的电子通道和减少活性物质的损失,以此来改善电池电化学性能。已经研究的电池正极结构主要包括核壳结构、蛋黄结构、层状多孔状结构、多孔球结构设计、网络拓扑结构设计、空心球结构设计、纤维状结构等。
金属化合物方面,金属氧化物和硫化物做正极材料也是研究的热点,而对金属氮化物的研究却很少。金属硫化物中,除了钛、钒和锆硫化物(TiS2、VS和ZrS2),硫化钴(CoS2和Co9S8)的双亲和性和金属性质使其作为正极材料也是很有前景的。
导电聚合物由于其良好的导电性质,同样受到了广泛关注。但是在多次循环后,导电聚合物的性质会明显下降,使其应用受到限制,现在的常规做法是将其和其他材料结合起来。但是导电聚合物的优越性质是不能忽视的,将其作为未来的研究方向也是很有前景的。
为了减少放电过程中活性物质损失,研究人员常常在正极材料中引入杂质原子或是特殊的官能团。氮和氧元素形成的官能团对多硫化物具有强烈的亲和力,常常作为杂质原子修饰正极材料。除了使用电负性原子与多硫化物中的锂结合之外,还可以使用正电性原子与多硫化物中的硫结合。对硼掺杂的碳材料的研究表明硼原子能够强烈地吸附锂多硫化物,这归因于B―S键的强相互作用119。将镍基金属有机骨架作为正极材料的研究报道同样证明了Ni―S键具有很强的相互作用120。最近关于Ti4O7和Ti2C的研究也证明了Ti―S键之间的强相互作用可以增强硫正极的循环性能。对此进行推测,其它正电性原子也有可能成为正极材料的掺杂元素。
7.2 负极材料
近几年关于硫正极改性的文献特别多,而对锂硫电池锂金属负极进行改性保护的文献却很少。目前对锂负极的保护常见的是使用硝酸锂作为电解质添加剂,保护锂金属负极不受溶解的多硫化物的影响。虽然硝酸锂是一种有效的添加剂,但其强氧化性能会带来严重的安全问题,特别是在高浓度和高温条件下121。此外,这种添加剂可能会限制电池的库仑效率(通常为90%至98%)。为了保持优异的稳定性和长的循环寿命,锂硫电池的库仑效率值需达99.9%及以上,显然硝酸锂作为电池添加剂需要进一步改进。目前,在锂金属负极的电镀-剥离可逆性上已经有了一定的进展122–125,包括使用电解质添加剂(例如氟化锂和六氟磷酸铯)以及改变负极保护结构(例如互连的中空碳球和氮化硼膜),希望通过形成稳定的SEI抑制枝晶生长。解决锂金属负极问题的另一种方法是使用其他材料替代完全锂化的负极,如上所述。但是这种电池锂离子的供应有限,在循环期间会造成活性物质不可逆的损失。此外,这种负极材料在循环过程中还会出现体积膨胀以及形成不稳定的SEI膜等问题,盲目替换负极材料将对循环性能有影响。为了在这个方向中有所突破,需要解决材料体积膨胀和不稳定的SEI问题。
7.3 其他重要组分
对正极材料进行改性有助于减轻多硫化物溶解的问题,但不能再根本上解决问题。因此,迫切需要一种不会明显溶解多硫化锂物质的新类型的电解质。液体电解质是锂硫电池常见的电解质类型,其具有高的离子电导率,但是存在易形成锂枝晶、多硫化合物的溶解与穿梭以及安全隐患问题。出于这些方面的考虑,研究人员开发出的固态电解质可以很好解决上述问题,但是固体电解质本身有离子电导率低和反应界面不稳定等问题。离子液体电解质也是一种十分有前途的锂硫电池电解质,离子液体电解质有阻燃、高离子电导率、宽的电化学稳定性窗口以及对负极锂的保护等优势,但是其存在负极的不可逆还原性、同多硫化合物反应产生副产品等缺点。因此合理选择功能性的电解质与正极材料保持协同作用是十分重要的。
隔膜和正极、电解质一样重要,改性的多功能隔膜通常通过浆料涂覆技术、真空过滤、磁控溅射或丝网印刷来预处理,以形成薄层涂层。浆料涂覆技术能够和传统的电池制造工艺相兼容,但是多出来的涂层不可避免地会增加隔膜厚度,增加电池的重量以及内部电阻。真空过滤能够使电池成分均匀分散,实现隔膜和电解质之间的紧密接触,然而,渗透过程通常需要投入很大成本购买设备,不利于工业大规模生产。磁控溅射能够很方便地制备多功能涂层,但是高能量的溅射离子和导电金属原子将增加沉积薄膜对下层的粘附力,可能会损坏电池隔膜。丝网印刷方法可以增加薄膜的厚度并控制其形状,但是该方法相当复杂并且相对昂贵。因此,研发重量轻、制备简单的新型物理/化学功能涂料可能是下一步锂硫电池的研究主流。最近的研究主要包括聚合物隔膜,纳米纤维隔膜和陶瓷隔膜等新型多功能隔膜,这些新型多功能隔膜具有离子选择性以及抑制多硫化物溶解的功能,引起了人们的广泛关注。
7.4 商业化
随着对锂硫电池正极材料的研究,公开发表的文献出现了很高的比容量以及长的循环寿命。但将锂硫电池投入实际应用还有很多方面要综合考虑。
电极中活性物质的含量。在迄今为止的文献中都是以活性材料硫的质量来计算比容量值的。没有考虑由其它电化学惰性材料提供的静重量,例如主体材料,碳添加剂和粘合剂(其通常占电极的40至60%,质量分数),同样的重量下,正极总共能提供的比能量是远远小于锂离子电池的。因此为了同锂离子电池进行竞争,增加硫电极中活性材料的百分比是关键。可以通过进一步提高正极材料的导电性,尽量减少碳添加剂。另一方面是在锂硫电池的电压范围内寻找具有电化学活性的电极材料,这些活性电体材料包括金属氧化物,氮化物和硫化物。
活性物质的面积质量负荷。目前,硫正极中活性材料的含量通常为1–3 mAh∙cm−2,而商业锂离子电池具有约为35 mAh∙cm−2。因此,为了将锂硫电池投入实际生产,必须确保正极具有较高的有效硫含量,这可以通过在电极上进行辊压以改进它们的整体颗粒与颗粒的接触,但目前并未达到预期的效果。制备较高硫含量的正极也意味着电池要在更高电流密度下进行循环反应,这是,锂金属负极的典型问题(包括枝晶生长和不稳定的SEI膜)在高电流密度下会被放大,锂金属负极的可逆性可能会因此遭到破坏。
电池性能的一致性。要想使锂硫电池实现大规模量产,就需要制定出统一的标准。但是目前主流文献中评判锂硫电池好坏的标准是放电比容量以及循环寿命,对工艺参数、制备成功率等参数均未提及。实际使用中并不需要容量无限接近硫理论放电容量的锂硫电池,在达到一定性能的前提下,能够保证性能的一致性是锂硫电池商业化成产的必要条件。
合成过程的简单性和相容性。为了实现锂-硫电池的商业化,合成过程需要简单且低成本。然而,文献中所述的许多步骤本质上是复杂的、昂贵的。寻求更为合理的生产途径也是其能够商业化的重要一环。
放电过程中的密封性及安全性。想要使消费者接受锂硫电池,除了同等价位锂硫电池具有比锂离子电池好得多的性能以外,安全性才是更为重要的。目前市面上的锂离子电池还经常出现燃烧爆炸现象,如何避免锂硫电池在反应过程中内部和空气的接触,避免电池过热发生爆炸现象,也是研究人员所要关注的重点。
8 结论
近年来,通过使用各种碳聚合物、导电聚合物等设计新型硫正极的文献数量迅速增加可以看出,人们对锂硫电池的研究从未中断。通过进一步的深入研究和优化设计,在不久的将来,锂硫电池一定能够实现商业化。
(1) Dahn, J. R.; Zheng, T.; Liu, Y.; Xue, J. S.1995,, 590. doi: 10.1126/science.270.5236.590
(2) Goodenough, J. B.; Park, K. S.2013,, 1167. doi: 10.1021/ja3091438
(3) Whittingham, M. S.1976,, 1126. doi: 10.1126/science.192.4244.1126
(4) Whittingham, M. S.2014,, 11414. doi: 10.1021/cr5003003
(5) Adelhelm, P.; Hartmann, P.; Bender, C. L.; Busche, M.; Eufinger, C.; Janek, J.2015,, 1016. doi: 10.3762/bjnano.6.105
(6) Bruce, P. G.; Freunberger, S. A.; Hardwick, L. J.; Tarascon, J. M.2012,, 19. doi: 10.1038/nmat3191
(7) Evers, S.; Nazar, L. F.2013,, 1135. doi: 10.1021/ar3001348
(8) Ma, L.; Hendrickson, K. E.; Wei, S.; Archer, L. A.2015,, 315. doi: 10.1016/j.nantod.2015.04.011
(9) Manthiram, A.; Fu, Y.; Chung, S. H.; Zu, C.; Su, Y. S.2014,, 11751. doi: 10.1021/cr500062v
(10) Yang, Y.; Zheng, G.; Cui, Y.2013,, 3018. doi: 10.1039/c2cs35256g
(11) Yin, Y. X.; Xin, S.; Guo, Y. G.; Wan, L. J.2013,, 13186. doi: 10.1002/anie.201304762
(12) Armand, M.; Tarascon, J. M.2008,, 652. doi: 10.1038/451652a
(13) Chiang, Y. M.2010,, 1485. doi: 10.1126/science.1198591
(14) Seh, Z. W.; Zhang, Q.; Li, W.; Zheng, G.; Yao, H.; Cui, Y.2013,, 3673. doi: 10.1039/c3sc51476e
(15) Zheng, G.; Zhang, Q.; Cha, J. J.; Yang, Y.; Li, W.; Seh, Z. W.; Cui, Y.2013,, 1265. doi: 10.1021/nl304795g
(16) Li, W.F.; Liu, M.N.; Wang, J.; Zhang, Y.G.2017,, 165. [李宛飞, 刘美男, 王 健, 张跃钢. 物理化学学报, 2017,, 165.] doi: 10.3866/PKU.WHXB201609232
(17) Qie, L.; Manthiram, A.2016,, 46. doi: 10.1021/acsenergylett.6b00033
(18) Han, S. C.; Song, M. S.; Lee, H.; Kim, H. S.; Ahn, H. J.; Lee, J. Y.2003,, A889. doi: 10.1149/1.1576766
(19) Yuan, L.; Yuan, H.; Qiu, X.; Chen, L.; Zhu, W.2009,, 1141. doi: 10.1016/j.jpowsour.2008.12.149
(20) Peng, H. J.; Hou, T. Z.; Zhang, Q.; Huang, J. Q.; Cheng, X. B.; Guo, M. Q.; Yuan, Z.; He, L. Y.; Wei, F.2014,. 1400227. doi: 10.1002/admi.201400227
(21) Lee, J. S.; Manthiram, A.2017,, 54. doi: 10.1016/j.jpowsour.2017.01.049
(22) Hwang, J. Y.; Kim, H. M.; Lee, S. K.; Lee, J. H.; Abouimrane, A.; Khaleel, M. A.; Belharouak, I.; Manthiram, A.; Sun, Y. K.2016,.1501480. doi: 10.1002/aenm.201501480
(23) Mao, Y.; Li, G.; Guo, Y.; Li, Z.; Liang, C.; Peng, X.; Lin, Z.2017,, 14628. doi: 10.1038/ncomms14628
(24) Fang, R.; Zhao, S.; Pei, S.; Qian, X.; Hou, P. X.; Cheng, H. M.; Liu, C.; Li, F.2016,, 8676. doi: 10.1021/acsnano.6b04019
(25) Wang, H.; Yang, Y.; Liang, Y.; Robinson, J. T.; Li, Y.; Jackson, A.; Cui, Y.; Dai, H.2011,, 2644. doi: 10.1021/nl200658a
(26) Ji, L.; Rao, M.; Zheng, H.; Zhang, L.; Li, Y.; Duan, W.; Guo, J.; Cairns, E. J.; Zhang, Y.2011,, 18522.
(27) Ji, X.; Nazar, L. F.2010,, 9821. doi: 10.1039/b925751a
(28) Chen, H.; Wang, C.; Dong, W.; Lu, W.; Du, Z.; Chen, L.2015,, 798. doi: 10.1021/nl504963e
(29) Ding, B.; Yuan, C.; Shen, L.; Xu, G.; Nie, P.; Lai, Q.; Zhang, X.2013,, 1096. doi: 10.1039/c2ta00396a
(30) Zhao, M. Q.; Zhang, Q.; Huang, J. Q.; Tian, G. L.; Nie, J. Q.; Peng, H. J.; Wei, F.2014,. 3410. doi: 10.1038/ncomms4410
(31) Wang, C.; Su, K.; Wan, W.; Guo, H.; Zhou, H.; Chen, J.; Zhang, X.; Huang, Y.2014,, 5018. doi: 10.1039/c3ta14921h
(32) Qiu, Y.; Li, W.; Zhao, W.; Li, G.; Hou, Y.; Liu, M.; Zhou, L.; Ye, F.; Li, H.; Wei, Z.; Yang, S.; Duan, W.; Ye, Y.; Guo, J.; Zhang, Y.2014,, 4821. doi: 10.1021/nl5020475
(33) Wang, Y.; Yan, Y. L.; Ren, B.; Yang, R.; Zhang, W.; Xu, Y. H.2017,, 012013. doi: 10.1088/1757-899X/182/1/012013
(34) Ye, H.; Yin, Y. X.; Xin, S.; Guo, Y. G.2013,, 6602. doi: 10.1039/c3ta10735c
(35) Park, M. S.; Jeong, B. O.; Kim, T. J.; Kim, S.; Kim, K. J.; Yu, J. S.; Jung, Y.; Kim, Y. J.2014,, 265. doi: 10.1016/j.carbon.2013.11.001
(36) He, J.; Chen, Y.; Lv, W.; Wen, K.; Xu, C.; Zhang, W.; Li, Y.; Qin, W.; He, W.2016,, 10981. doi: 10.1021/acsnano.6b05696
(37) Li, J.; Guo, J.; Deng, J.; Huang, Y.2017,, 188. doi: 10.1016/j.matlet.2016.12.012
(38) Wei, H.; Rodriguez, E. F.; Best, A. S.; Hollenkamp, A. F.; Chen, D.; Caruso, R. A.2016,, 1601616. doi: 10.1002/aenm.201601616
(39) Zhang, Y.; Wang, L.; Zhang, A.; Song, Y.; Li, X.; Feng, H.; Wu, X.; Du, P.2010,, 835. doi: 10.1016/j.ssi.2010.04.010
(40) Ponraj, R.; Kannan, A. G.; Ahn, J. H.; Kim, D. W.2016,, 4000. doi: 10.1021/acsami.5b11327
(41) Tao, Y. Q.; Wei, Y.; Liu, Y.; Wang, J.; Qiao, W.; Ling, L.; Long, D.2016,, 3230. doi: 10.1039/c6ee01662f
(42) Faber, M. S.; Lukowski, M. A.; Ding, Q.; Kaiser, N. S.; Jin, S.2014,, 21347. doi: 10.1021/jp506288w
(43) Dirlam, P. T.; Park, J.; Simmonds, A. G.; Domanik, K.; Arrington, C. B.; Schaefer, J. L.; Oleshko, V. P.; Kleine, T. S.; Char, K.; Glass, R. S.; Soles, C. L.; Kim, C.; Pinna, N.; Sung, Y. E.; Pyun, J.2016,, 13437. doi: 10.1021/acsami.6b03200
(44) Liu, Z.; Zheng, X.; Luo, S. L.; Xu, S. Q.; Yuan, N. Y.; Ding, J. N.2016,, 13395. doi: 10.1039/c6ta05635k
(45) Lei, T.; Chen, W.; Huang, J.; Yan, C.; Sun, H.; Wang, C.; Zhang, W.; Li, Y.; Xiong, J.2016,1601843. doi: 10.1002/Aenm.201601843
(46) Zhang, S. S.; Tran, D. T.2016,, 4371. doi: 10.1039/c6ta01214k
(47) Cai, K.; Song, M.K.; Cairns, E. J.; Zhang, Y.2012,, 6474. doi: 10.1021/nl303965a
(48) Liu, J. D.; Zheng, X. S.; Shi, Z. F.; Zhang, S. Q.2014,, 659. doi: 10.1007/s11581-013-1019-6
(49) Sun, K.; Su, D.; Zhang, Q.; Bock, D. C.; Marschilok, A. C.; Takeuchi, K. J.; Takeuchi, E. S.; Gan, H.2015,, A2834. doi: 10.1149/2.1021514jes
(50) Chen, L.; Liu, J. D.; Zhang, S. Q.2013,, 1127. doi: 10.3724/Sp.J.1077.2013.13017
(51) Li, X.; Lu, Y.; Hou, Z.; Zhang, W.; Zhu, Y.; Qian, Y.; Liang, J.; Qian, Y.2016,, 19550. doi: 10.1021/acsami.6b06565
(52) Lu, Y.; Li, X.; Liang, J.; Hu, L.; Zhu, Y.; Qian, Y.2016,, 17616. doi: 10.1039/c6nr05626a
(53) Cui, Z.; Zu, C.; Zhou, W.; Manthiram, A.; Goodenough, J. B.2016,, 6926. doi: 10.1002/adma.201601382
(54) Hao, Z.; Yuan, L.; Chen, C.; Xiang, J.; Li, Y.; Huang, Z.; Hu, P.; Huang, Y.2016,, 17711. doi: 10.1039/c6ta07411a
(55) Liang, X.; Garsuch, A.; Nazar, L. F.2015,, 3907. doi: 10.1002/anie.201410174
(56) Peng, H. J.; Zhang, G.; Chen, X.; Zhang, Z. W.; Xu, W. T.; Huang, J. Q.; Zhang, Q.2016,, 12990. doi: 10.1002/ange.201605676
(57) Al Salem, H.; Chitturi, V. R.; Babu, G.; Santana, J. A.; Gopalakrishnan, D.; Arava, L. M. R.2016,, 110301. doi: 10.1039/c6ra22434b
(58) Wang, J.; Chen, J.; Konstantinov, K.; Zhao, L.; Ng, S. H.; Wang, G. X.; Guo, Z. P.; Liu, H. K.2006,, 4634. doi: 10.1016/j.electacta.2005.12.046
(59) Wu, F.; Chen, J.; Chen, R.; Wu, S.; Li, L.; Chen, S.; Zhao, T.2011,, 6057. doi: 10.1021/jp1114724
(60) Xiao, L.; Cao, Y.; Xiao, J.; Schwenzer, B.; Engelhard, M. H.; Saraf, L. V.; Nie, Z. M.; Exarhos, G. J.; Liu, J.2012,, 1176. doi: 10.1002/adma.201103392
(61) Zhou, W.; Yu, Y.; Chen, H.; DiSalvo, F. J.; Abruna, H. D.2013,, 16736. doi: 10.1021/ja409508q
(62) Li, W.; Zhang, Q.; Zheng, G.; Seh, Z. W.; Yao, H.; Cui, Y.2013,, 5534. doi: 10.1021/nl403130h
(63) Yan, M.; Zhang, Y.; Li, Y.; Huo, Y.; Yu, Y.; Wang, C.; Jin, J.; Chen, L.; Hasan, T.; Wang, B.; Su, B. L.2016,, 9403. doi: 10.1039/c6ta03211g
(64) Ji, X.; Lee, K. T.; Nazar, L. F.2009,, 500. doi: 10.1038/nmat2460
(65) Zhang, C.; Wu, H. B.; Yuan, C.; Guo, Z.; Lou, X. W.2012,, 9730. doi: 10.1002/ange.201205292
(66) Kim, S.; Jung, Y.; Lim, H. S.2004,, 889. doi: 10.1016/j.electacta.2004.01.093
(67) Jin, B.; Kim, J. U.; Gu, H. B.2003,, 148. doi: 10.1016/S0378-7753(03)00113-7
(68) Barchasz, C.; Lepretre, J. C.; Patoux, S.; Alloin, F.2013,, A430. doi: 10.1149/2.022303jes
(69) Azimi, N.; Weng, W.; Takoudis, C.; Zhang, Z.2013,, 96. doi: 10.1016/j.elecom.2013.10.020
(70) Lu, H.; Zhang, K.; Yuan, Y.; Qin, F.; Zhang, Z.; Lai, Y.; Liu, Y.2015,, 55. doi: 10.1016/j.electacta.2015.02.031
(71) Xu, R.; Belharouak, I.; Li, J. C. M.; Zhang, X.; Bloom, I.; Bareno, J.2013,, 833. doi: 10.1002/aenm.201200990
(72) Aurbach, D.; Pollak, E.; Elazari, R.; Salitra, G.; Kelley, C. S.; Affinito, J.2009,, A694. doi: 10.1149/1.3148721
(73) Zhu, X.; Wen, Z.; Gu, Z.; Lin, Z.2005,, 269. doi: 10.1016/j.jpowsour.2004.07.002
(74) Liang, X.; Wen, Z.; Liu, Y.; Zhang, H.; Huang, L.; Jin, J.2011,, 3655. doi: 10.1016/j.jpowsour.2010.12.052
(75) Teran, A. A.; Balsara, N. P.2011,, 9267. doi: 10.1021/ma202091z
(76) Hayashi, A.; Ohtomo, T.; Mizuno, F.; Tadanaga, K.; Tatsumisago, M.2003,, 701. doi: 10.1016/S1388-2481(03)00167-X
(77) Machida, N.; Kobayashi, K.; Nishikawa, Y.; Shigematsu, T.2004,, 247. doi: 10.1016/j.ssi.2003.11.033
(78) Agostini, M.; Aihara, Y.; Yamada, T.; Scrosati, B.; Hassoun, J.2013,, 48. doi: 10.1016/j.ssi.2013.04.024
(79) Xu, R. C.; Xia, X. H.; Wang, X. L.; Xia, Y.; Tu, J. P.2017,, 2829. doi: 10.1039/c6ta10142a
(80) Wang, Q.; Wen, Z.; Jin, J.; Guo, J.; Huang, X.; Yang, J.; Chen, C.2016,, 1637. doi: 10.1039/c5cc08279j
(81) Yan, Y.; Yin, Y. X.; Xin, S.; Su, J.; Guo, Y. G.; Wan, L. J.2013,, 58. doi: 10.1016/j.electacta.2012.12.077
(82) Wang, L. N.; Liu, J.; Yuan, S.; Wang, Y.; Xia, Y.2016,, 224. doi: 10.1039/c5ee02837j
(83) Park, J. W.; Yamauchi, K.; Takashima, E.; Tachikawa, N.; Ueno, K.; Dokko, K.; Watanabe, M.2013,, 4431. doi: 10.1021/jp400153m
(84) Wu, F.; Zhu, Q.; Chen, R.; Chen, N.; Chen, Y.; Ye, Y.; Qian, J.; Li, L.2015,, 10. doi: 10.1016/j.jpowsour.2015.07.033
(85) Chen, Z. J.; Xue, T.; Lee, J. M.2012,, 10564. doi: 10.1039c2ra21772d
(86) Wang, L. N.; Byon, H. R.2013,, 207. doi: 10.1016/j.jpowsour.2013.02.068
(87) Bai, S.; Liu, X.; Zhu, K.; Wu, S.; Zhou, H.2016,16094. doi: 10.1038/NENERGY.2016.94
(88) Gu, M.; Lee, J.; Kim, Y.; Kim, J. S.; Jang, B. Y.; Lee, K. T.; Kim, B. S.2014,, 46940. doi: 10.1039/c4ra09718a
(89) Chung, S. H.; Han, P.; Singhal, R.; Kalra, V.; Manthiram, A.2015,. 1500738. doi: 10.1002/aenm.201500738
(90) Ma, G.; Huang, F.; Wen, Z.; Wang, Q.; Hong, X.; Jin, J.; Wu, X.2016,, 16968. doi: 10.1039/C6TA07198H
(91) Nair, J. R.; Bella, F.; Angulakshmi, N.; Stephan, A. M.; Gerbaldi, C.2016,, 69. doi: 10.1016/j.ensm.2016.01.008
(92) Chang, C. H.; Chung, S. H.; Manthiram, A.2015,, 18829. doi: 10.1039/c5ta05053g
(93) Peng, H. J.; Zhang, Q.2015,, 11018. doi: 10.1002/anie.201505444
(94) Song, J.; Gordin, M. L.; Xu, T.; Chen, S.; Yu, Z.; Sohn, H.; Lu, J.; Ren, Y.; Duan, Y.; Wang, D.2015,, 4325. doi: 10.1002/anie.201411109
(95) Fan, C. Y.; Yuan, H. Y.; Li, H. H.; Wang, H. F.; Li, W. L.; Sun, H. Z.; Wu, X. L.; Zhang, J. P.2016,, 16108. doi: 10.1021/acsami.6b04578
(96) Hou, T. Z.; Chen, X.; Peng, H. J.; Huang, J. Q.; Li, B. Q.; Zhang, Q.; Li, B.2016,, 3283. doi: 10.1002/smll.201600809
(97) Zeng, F.; Jin, Z.; Yuan, K.; Liu, S.; Cheng, X.; Wang, A.; Wang, W.; Yang, Y. S.2016,, 12319. doi: 10.1039/c6ta02680j
(98) Lin, Z.; Liang, C.2015,, 936. doi: 10.1039/c4ta04727c
(99) Zhang, Z.; Lai, Y.; Zhang, Z.; Li, J.2015,, 166. doi: 10.1016/j.ssi.2015.06.018
(100) Zhu, J.; Ge, Y.; Kim, D.; Lu, Y.; Chen, C.; Jiang, M.; Zhang, X.2016,, 176. doi: 10.1016/j.nanoen.2015.12.022
(101) Cheng, X.; Wang, W.; Wang, A.; Yuan, K.; Jin, Z.; Yang, Y.; Zhao, X.2016,, 89972. doi: 10.1039/c6ra14581g
(102) Yao, H.; Yan, K.; Li, W.; Zheng, G.; Kong, D.; Seh, Z. W.; Narasimhan, V. K.; Liang, Z.; Cui, Y.2014,, 3381. doi: 10.1039/c4ee01377h
(103) Chung, S. H.; Manthiram, A.2014,, 7352. doi: 10.1002/adma.201402893
(104) Kim, J. S.; Hwang, T. H.; Kim, B. G.; Min, J.; Choi, J. W.2014,, 5359. doi: 10.1002/adfm.201400935
(105) Cheng, X. B.; Hou, T. Z.; Zhang, R.; Peng, H. J.; Zhao, C. Z.; Huang, J. Q.; Zhang, Q.2016,, 2888. doi: 10.1002/adma.201506124
(106) Han, Y.; Duan, X.; Li, Y.; Huang, L.; Zhu, D.; Chen, Y.2015,, 160. doi: 10.1016/j.materresbull.2015.03.042
(107) Liang, X.; Wen, Z.; Liu, Y.; Wu, M.; Jin, J.; Zhang, H.; Wu, X.2011,, 9839. doi: 10.1016/j.jpowsour.2011.08.027
(108) Wu, M.; Jin, J.; Wen, Z.. 2016,, 40270. doi: 10.1039/c6ra05316e
(109) Liu, S.; Li, G. R.; Gao, X. P.2016,, 7783. doi: 10.1021/acsami.5b12231
(110) Wang, X.; Hou, Y.; Zhu, Y.; Wu, Y.; Holze, R.2013,1401. doi: 10.1038/srep01401.
(111) Ma, G.; Wen, Z.; Wang, Q.; Shen, C.; Jin, J.; Wu, X.2014,, 19355. doi: 10.1039/c4ta04172k
(112) Luo, J.; Lee, R. C.; Jin, J. T.; Weng, Y. T.; Fang, C. C.; Wu, N. L.2017,, 963. doi: 10.1039/c6cc09248a.
(113) Zhang, Y. J.; Xia, X. H.; Wang, X. L.; Gu, C. D.; Tu, J. P.2016,, 66161. doi: 10.1039/c6ra13039a
(114) Brückner, J.; Thieme, S.; Grossmann, H. T.; Dörfler, S.; Althues, H.; Kaskel, S.2014,, 82. doi: 10.1016/j.jpowsour.2014.05.143
(115) Zhang, X.; Wang, W.; Wang, A.; Huang, Y.; Yuan, K.; Yu, Z.; Qiu, J.; Yang, Y.2014,, 11660. doi: 10.1039/c4ta01709a
(116) Hassoun, J.; Scrosati, B.2010,, 2371. doi: 10.1002/anie.200907324
(117) Huang, C.; Xiao, J.; Shao, Y.; Zheng, J.; Bennett, W. D.; Lu, D.; Saraf, L. V.; Engelhard, M.; Ji, L. W.; Zhang, J.; Li, X.; Graff, G. L.; Liu, J.2014,,3015. doi: 10.1038/Ncomms4343
(118) Ma, G.; Wen, Z.; Wang, Q.; Shen, C.; Jin, J.; Wu, X.2014,, 19355. doi: 10.1039/c4cc05535g
(119) Yang, C. P.; Yin, Y. X.; Ye, H.; Jiang, K. C.; Zhang, J.; Guo, Y. G.2014,, 8789. doi: 10.1021/am501627f
(120) Zheng, J.; Tian, J.; Wu, D.; Gu, M.; Xu, W.; Wang, C.; Gao, F.; Engelhard, M. H.; Zhang, J. G.; Liu, J.; Xiao, J.2014,, 2345. doi: 10.1021/nl404721h
(121) Zhang, S. S.2013,, 153. doi: 10.1016/j.jpowsour.2012.12.102
(122) Aurbach, D.; Zinigrad, E.; Cohen, Y.; Teller, H.2002,, 405. doi: 10.1016/S0167-2738(02)00080-2
(123) Park, M. S.; Ma, S. B.; Lee, D. J.; Im, D.; Doo, S. G.; Yamamoto, O.2014,3815. doi: 10.1038/srep03815
(124) Zhang, Y.; Qian, J.; Xu, W.; Russell, S. M.; Chen, X.; Nasybulin, E.; Bhattacharya, P.; Engelhard, M. H.; Mei, D.; Cao, R.; Ding, F.; Cresce, A. V.; Xu, K.; Zhang, J. G.2014,, 6889. doi: 10.1021/nl5039117
(125) Zheng, G.; Lee, S. W.; Liang, Z.; Lee, H. W.; Yan, K.; Yao, H.; Wang, H.; Li, W.; Chu, S.; Cui, Y.2014,, 618. doi: 10.1038/nnano.2014.152
Advances in High-Performance Lithium-Sulfur Batteries
LIU Shuai YAO Lu ZHANG Qin LI Lu-Lu HU Nan-Tao WEI Liang-Ming WEI Hao*
()
Lithium-sulfur batteries are considered to be rather latest and high-performance storage batteries due to their high theoretical specific capacity (1675 mAh∙g−1), high energy density (2600 Wh∙kg−1), environmental friendliness, low cost, and safety. These features make them important in the field of mobile electric vehicles and portable devices. However, because of rapid capacity attenuation with poor cycle and rate performances, these batteries are far from ideal for commercial applications. This paper reviews the entire and latest studies in lithium-sulfur batteries. Cathodes, electrolyte, separators, and anodes protection are introduced in detail. The existing lithium-sulfur batteries defects and problems are analyzed. Finally, we provide some insights into the future direction and prospects of lithium batteries.
Lithium-sulfur battery; Cathode; Anode protection; Separator; Application
April 13, 2017;
May 26, 2017;
June 2, 2017.
Corresponding author. Email: haowei@sjtu.edu.cn; Tel: +86-21-34204322.
10.3866/PKU.WHXB201706021
O649
The project was supported by the National Natural Science Foundation of China (61376003), the Shanghai Pujiang Program, China (16PJD027), and the Medical-Engineering Crossover Fund of Shanghai Jiao Tong University, China (YG2015MS23, YG2016MS71).
国家自然科学基金(61376003),上海市浦江人才计划(16PJD027),上海交通大学医工交叉研究基金(YG2015MS23, YG2016MS71)资助项目
猜你喜欢
无机化学学报(2022年10期)2022-10-10
科学技术创新(2021年26期)2021-09-15
当代水产(2021年3期)2021-07-20
装备制造技术(2020年4期)2020-12-25
中国新技术新产品(2019年2期)2019-04-12
浙江化工(2018年1期)2018-02-03
中南大学学报(自然科学版)(2016年2期)2017-01-19
中国资源综合利用(2016年7期)2016-02-03
橡胶工业(2015年4期)2015-02-23
应用化工(2014年11期)2014-08-16
