
Effect of Surrounding Media on Ultrafast Plasmon Dynamics of Gold Nanoparticles
2018-01-12 06:07CHENXiaoYuWANGJingDongYUAnChi
物理化学学报 2017年11期
CHEN Xiao-Yu WANG Jing-Dong YU An-Chi
Effect of Surrounding Media on Ultrafast Plasmon Dynamics of Gold Nanoparticles
CHEN Xiao-Yu WANG Jing-Dong YU An-Chi*
()
Herein, we prepared four samples, namely gold/poly(sodium--styrenesulfonate) (Au/PSS), gold/silicon dioxide (Au/SiO2), gold/titanium dioxide (Au/TiO2), and gold/cuprous oxide (Au/Cu2O) core/shell nanocomposites, to investigate how the surrounding medium affects the ultrafast plasmon dynamics of Au nanoparticles (NPs). We recorded femtosecond transient absorption spectra of Au NPs in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O core/shell nanocomposites at various time delays. We found that the spectral features in the femtosecond transient absorption spectra of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites were dramatically different from those of Au NPs in Au/PSS and Au/SiO2core/shell nanocomposites. A comprehensive analysis of the ultrafast plasmon dynamics of Au NPs in the core/shell nanocomposites revealed that following excitation of the resonance plasmon band of Au NPs, the exited electrons could be efficiently transferred into the conduction bands of TiO2and Cu2O in Au/TiO2and Au/Cu2O core/shell nanocomposites.
Femtosecond transient absorption spectroscopy; Plasmon resonance excitation; Electron transfer; Gold nanoparticle; Core/shell structure
1 Introduction
Gold nanoparticles (Au NPs) have been extensively investigated due to their potential applications in sensing, photovoltaics, catalysis, imaging, and photothermal therapy1–7. Many of these applications are attributed to their unique optical properties. The main optical signature of Au NPs is its localized surface plasmon resonance (LSPR) absorption band which arises from the collective oscillations of the conduction band electrons of Au. The position of the LSPR absorption band of Au NPs in aqueous solution is usually centered at around 520 nm. However, the position of the LSPR absorption band of Au NPs can be tuned by changing its particle size or varying its surrounding medium8–10.
Femtosecond transient absorption spectroscopy has been widely used to investigate the electron relaxation dynamics of Au NPs8–21. Several processes, including electron-electron, electron-phonon, and phonon-phonon interactions, can contribute to the relaxation dynamics of Au NPs. Smith.11studied the electron relaxation dynamics of 15 nm Au NPs in water and observed time constants of 7 and 400 ps for the electron-phonon and phonon-phonon couplings. Perner.12measured the relaxation dynamics of 30 nm Au NPs embedded in a sol-gel matrix and found relaxation times of 4 and 200 ps. Inouye.13recorded the relaxation dynamics of 7.6 nm Au NPs in a SiO2glass matrix and found time constants of 2.8 and 120 ps for its electron-phonon and phonon-phonon relaxation processes. Link.14studied the dynamics of 15 nm Au NPs embedded in MgSO4powder and obtained time constants of 8 and 200 ps for its electron-phonon and phonon-phonon relaxation processes. It is obvious that the variation of the surrounding medium can change the relaxation dynamics of Au NPs after excitation. However, there is no systematical investigation on how the surrounding environment affects the ultrafast plasmon dynamics of Au NPs in the literature.
Besides, it is reported that the semiconductor-Au nanocomposites are of great interest for plasmon-induced photocatalytic applications such as the production of hydrogen, the reduction of thiocyanate, and the oxidation of carbon monoxide22–25. Several reports have shown that the excited electron of Au NPs after an excitation on its resonance plasmon band can be efficiently transferred into the conduction band of semiconductor nanocrystals22,26–28. Furube.27directly observed plasmon induced electron transfer from 10 nm gold nanodots to TiO2nanoparticles by using femtosecond transient absorption spectroscopy with an IR probe. Wu.28studied the plasmon-exciton interaction mechanisms in CdS/Au colloidal quantum confined plexcitonic nanorod heterostructures by transient absorption spectroscopy and found optical excitation of plasmons in the Au tip can lead to hot electron injection into the CdS rod.
To understand how the surrounding medium affect the ultrafast plasmon dynamics of Au NPs and whether the excited electron of Au NPs after its resonance plasmon band excitation can be transferred into the conduction band of a semiconductor, in this contribution, we synthesized four different Au/M core/shell nanocomposites, where M is poly(sodium-- styrenesulfonate) (PSS), silicon dioxide (SiO2), titanium dioxide (TiO2), and coprous oxide (Cu2O), respectively, and systematically recorded their respectively femtosecond transient absorption spectra at various time delays. We found that the feature of the femtosecond transient absorption spectra of Au NPs in Au/TiO2core/shell nanocomposite is dramatically different with that in Au/PSS and Au/SiO2core/shell nanocomposites, but it is quite similar as that in Au/Cu2O core/shell nanocomposite.
2 Experimental section
2.1 Materials
Hexadecyl trimethyl ammonium bromide (CTAB), sodium borohydride (NaBH4), L (+)-ascorbic acid, Poly(sodium--styrenesulfonate) (PSS) were purchased from Sigma, USA. Chloroauric acid tetrahydrate (HAuCl4∙4H2O), copper nitrate trihydrate (Cu(NO3)2∙3H2O), polyvinyl pyrrolidone (PVP K30), hydrazine hydrate (N2H4∙H2O), sulfuric acid (H2SO4), silver nitrate (AgNO3), titanium trichloride (TiCl3), hydrochloric acid (HCl), sodium bicarbonate (NaHCO3), 2-propanol (2-C3H7OH), ammonium hydroxide (NH3∙H2O), tetraethylorthosilicate (TEOS), sodium chloride (NaCl), ethanol were purchased from Sinopharm Chemical Reagent (China). All the reagents were of analytical grade and don’t need further purification. The details for the synthesizing procedure of Au/PSS core/shell composite, Au/SiO2core/shell composite, Au/TiO2core/shell composite, and Au/Cu2O core/shell composite were provided in the Supporting information.
2.2 Measurements
The morphologies and sizes of the samples were characterized by a Hitachi H-7650B transmission electron microscopy (TEM, Japan). The steady-state absorption spectra of the samples were recorded on a Varian Cary-50 UV-Visible spectrometer. The femtosecond transient absorption spectra of the samples were recorded on a homemade femtosecond transient absorption spectrometer29,30. Briefly, the outputs of a Spectra Physics 1 kHz amplified Ti:sapphire laser were used to pump an optical parametric amplifier (OPA) and to generate the white light continuum, respectively. The outputs of the OPA were used as the pump pulses, and the white light continuum generated by a spinning fused silica disk were used as the probe pulses. The pump and probe beams were non-collinearly focused into the sample cell using two achromatic lenses (300 mm focal length for pump and 100 mm focal length for probe, respectively). At the sample position, the average pump powers were about 0.2 mW. The timing between the pump and probe pulses was controlled using a Newport M-ILS250CC motorized translation stage. The time resolution of this setup was about 150 fs. In the femtosecond transient absorption spectroscopy measurement, the concentrations of the samples were adjusted to have an optical density of about 0.2 at their plasmon absorption maxima in a 1 mm pathlength sample cell. A home-made magnet stirring bar was placed inside the sample cell and rotated by an external magnet motor to keep the sample solution fresh. All samples except Au/Cu2O core/shell composite were suspended in water during the experiment measurement. The Au/Cu2O core/shell composite was dispersed in ethanol during the measurement. No protective agents were added for all the sample solutions.
3 Results and discussion
3.1 Steady-state absorption spectra of Au NPs in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O core/shell nanocomposites
To investigate the interaction between Au NPs and its surrounding medium, we respectively recorded the steady-state absorption spectrum of Au NPs in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O core/shell nanocomposites, as shown in Fig.1(A). The plasmon resonance of Au NPs in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O core/shell nanocomposites are respectively located at 529, 532, 544 and 579 nm, which is red-shifted as the shell medium is changed from PSS to SiO2, TiO2and Cu2O. It is known that the position of the plasmon absorption band of Au NPs can be tuned by changing its particle size or varying the dielectric constant of its surrounding medium8–10. The TEM measurements (Fig.S1 of the Supporting information) show that the averaged sizes of Au NPs in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O core/shell nanocomposites are quite similar and all about (40 ± 10) nm. Thus, the red shift for the plasmon resonance of Au NPs upon changing its environment is attributed to the variation of the dielectric constant of its surrounding medium.
Fig.1(B) shows the calculated absorption spectrum of Au NPs using the dipole approximation of the Mie theory14:

(1)
The values of the medium dielectric constant (m) were chosen and assumed to be frequency-independent as to match the observed plasmon resonance maxima.
3.2 Femtosecond transient absorption spectra of Au NPs in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O core/shell nanocomposites
Fig.2 shows the femtosecond transient absorption spectra of Au NPs in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O core/shell nanocomposites at various time delays. The feature of the femtosecond transient absorption spectra of Au NPs in Au/PSS and Au/SiO2core/shell nanocomposites (Fig.2(A, B)) are very similar as the earlier reports for Au NPs in solution, which consist of one negative absorption band and two positive absorption bands15–17. The negative absorption band has been attributed to laser induced plasmon band bleaching and the two positive absorption bands are commonly thought to be arisen from the heating of the plasmon resonance band after laser excitation. However, the features of the femtosecond transient absorption spectra of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites (Fig.2(C, D)) are dramatically different with those in Au/PSS and Au/SiO2core/shell nanocomposites (Fig.2(A, B)).
In the femtosecond transient absorption spectra of Au NPs in Au/PSS and Au/SiO2core/shell nanocomposites, the intensity of the positive absorption band at the blue side of the plasmon bleaching band is almost equal to the intensity of the positive absorption band at the red side of the plasmon bleaching band. However, in the femtosecond transient absorption spectra of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites, the intensity of the positive absorption band at the blue side of the plasmon bleaching band is obviously larger than the intensity of the positive absorption band at the red side of the plasmoning bleach band. Moreover, from the femtosecond transient absorption spectra shown in Fig.2, it is further found that the peak positions of the plasmoning bleach band of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites are shifted to blue as the increase of the time delay and that the peak positions of the plasmon bleaching band of Au NPs in Au/PSS and Au/SiO2core/shell nanocomposites do not change as the increase of the time delay. It is reported that the blue shift of the transient bleaching plasmon band as the increase of the time delay is a result of a special interaction between Au NPs and its surrounding medium32. Thus, both the blue shift of the plasmon bleaching band of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites as the increase of the time delay and the less intensity of the positive absorption band at the red side of the plasmon bleaching band of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites suggest that some interactions between Au and TiO2and between Au and Cu2O exist in Au/TiO2and Au/Cu2O core/shell nanocomposites, which is consistent with our steady-state absorption spectroscopy measurements where the line width of the plasmon absorption band of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites are much broader than those in Au/PSS and Au/SiO2core/shell nanocomposites. Aiboushev.32investigated the femtosecond transient absorption spectrum of Au NPs in TiO2mesoporous film and found a similar result as our current study for Au NPs in Au/TiO2core/shell nanocomposite. Karam.33studied the femtosecond transient absorption spectrum of colloidal TiO2-Au nanocomposites and also found a similar result as our current study for Au NPs in Au/TiO2core/shell nanocomposite. The femtosecond transient absorption spectra of Au NPs in Au/Cu2O core/shell nanocomposite has not been reported in the literature. In Karam’s study, the positive absorption band at the blue side of the plasmon bleaching band of Au NPs has been assigned to the contribution of theinterband transitions from the Auband to the Auband above Fermi level33.
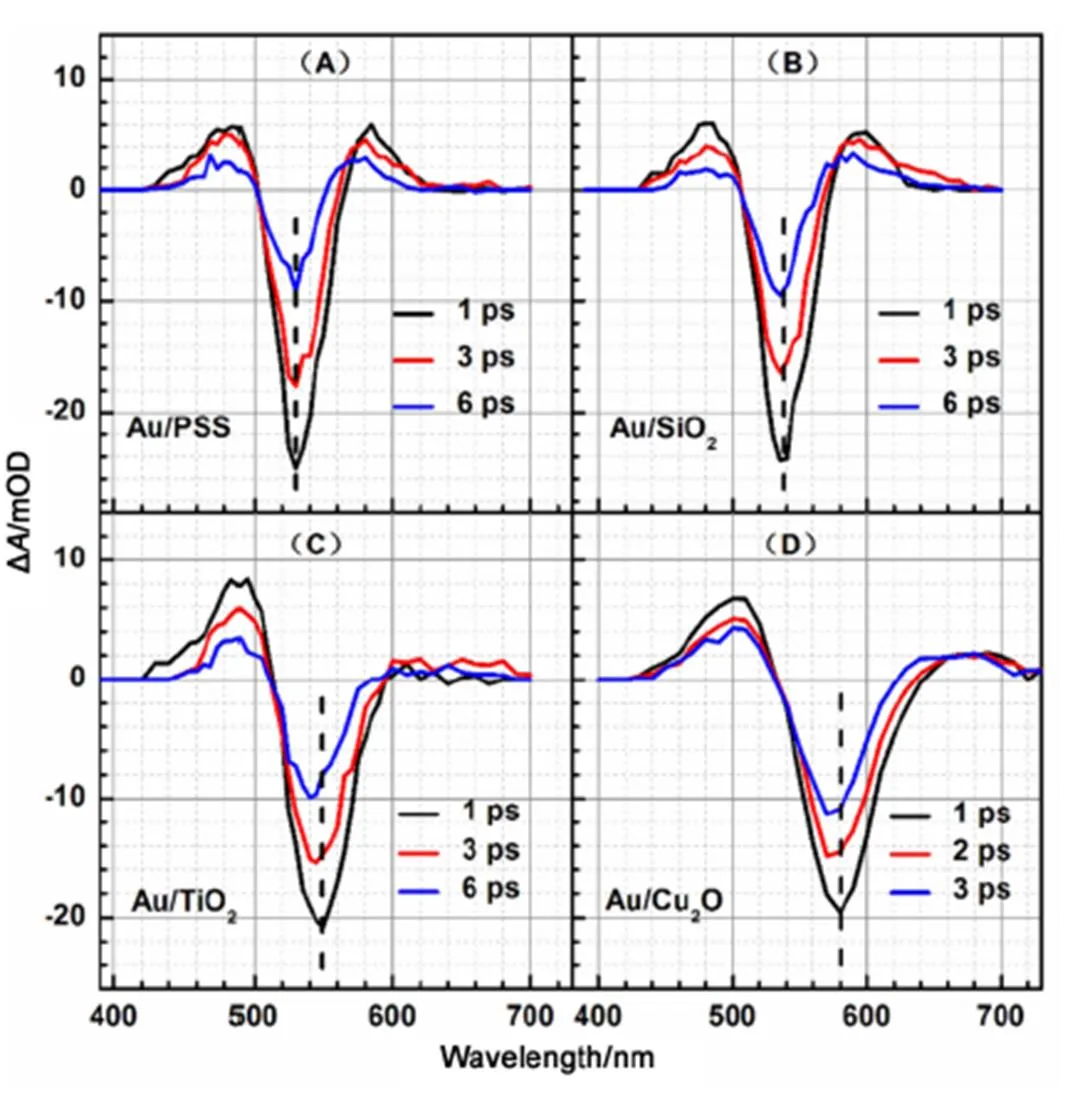
Fig.2 Femtosecond transient absorption spectra of Au NPs in Au/PSS (A), Au/SiO2 (B), Au/TiO2 (C), and Au/Cu2O (D) core/shell nanocomposites at various time delays.
3.3 Plasmon bleaching recovery kinetics of Au NPs in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O core/shell nanocomposites
To understand the interaction between Au NPs and its surrounding medium, we respectively recorded the plasmon bleaching recovery kinetic traces of Au NPs in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O core/shell nanocomposites, as shown in Fig.3. All the kinetic curves shown in Fig.3 can be fitted with a summation of two exponential decay functions and all the fitting parameters are summarized in Table 1. It is obvious that the time constant1listed in Table 1 changes with the variation of the surrounding medium, but that the time constant2listed in Table 1 maintains a constant within the experimental error.

Fig.3 Plasmon bleaching recovery kinetic traces of Au NPs in Au/PSS (A), Au/SiO2 (B), Au/TiO2 (C), and Au/Cu2O (D) core/shell nanocomposites.
The red lines are the fits. The pump/probe wavelengths for Au NPs in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O nanocomposites are 520/530, 520/540, 525/550, and 545/580 nm, respectively.
The1and2components listed in Table 1 have been respectively assigned to the electron-phonon and phonon-phonon scattering of Au NPs after laser excitation. The obtained1value for Au NPs in Au/PSS is similar as the earlier report where they measured the relaxation dynamics of 30 nm Au NPs embedded in a sol-gel matrix12. Besides, from the data listed in Table 1, it is further found that (1) the1value for Au NPs in Au/SiO2core/shell nanocomposite is almost equal to that for Au NPs in Au/PSS core/shell nanocomposite, (2) the1value for Au NPs in Au/TiO2core/shell nanocomposite is obviously longer than that for Au NPs in Au/PSS core/shell nanocomposite, and (3) the1value for Au NPs in Au/Cu2O core/shell nanocomposite is obviously shorter than that for Au NPs in Au/PSS core/shell nanocomposite. In general, the electron-phonon scattering of Au NPs is independent of its size and shape10. Thus, the changes of the electron-phonon scattering of Au NPs in Au/PSS, Au/SiO2, Au/TiO2and Au/Cu2O core/shell nanocomposites could be attributed to the variation of its surrounding medium.
Besides, it is reported that the excited electron of Au NPs after an excitation on its resonance plasmon band can be efficiently transferred into the conduction band of semiconductor nanocrystals (~200 fs)27. Both TiO2and Cu2O are semiconductors. Therefore, the obtained different1value for Au NPs in Au/TiO2and Au/Cu2O core/shell nanocompositescould also be understood through the electron transfer mechanism. Once the plasmon absorption of Au NPs in Au/TiO2(or Au/Cu2O) core/shell nanocomposite is excited, the excited electron can be immediately generated on the plasmon band of Au NPs and efficiently transferred into the conduction band of TiO2(or Cu2O) with hundreds of femtoseconds. As a consequence, the density of the electron on the Fermi level of Au NPs in Au/TiO2(or Au/Cu2O) core/shell nanocomposite is decreased upon light excitation. Then, the electron density decrease on the Fermi level of Au NPs could make the interband transition from the Auband to the Auband above Fermi level more easily in Au/TiO2(or Au/Cu2O) core/shell nanocomposite. In Fig.2, we observed that the femtosecond transient absorption features of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites are dramatically different with those of Au NPs in Au/PSS and Au/SiO2core/shell nanocomposites. The contribution of the positive absorption band at the blue side of plasmon bleaching band is more intense than the plasmon heating contribution (red side of plasmon bleaching band) in the femtosecond transient absorption spectra of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites. Thus, it is reasonable to suggest that the positive absorption bands at the blue side of plasmon bleaching band of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites are the interband transitions from the Auband to the Auband above Fermi level. As a consequence, the obtained time constant1for Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites are the electron recombination times from the conduction band of TiO2and Cu2O to the Fermi level of Au NPs. The obtained different value of1for Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites further indicated that the electron recombination rate from the conduction band of TiO2to the Fermi level of Au NPs in Au/TiO2core/shell nanocomposite is different with that from the conduction band of Cu2O to the Fermi level of Au NPs in Au/Cu2O core/shell nanocomposite. This also suggested that the obtained1components for Au NPs in Au/PSS and Au/SiO2core/shell nanocomposites are the electron-phonon coupling times of Au NPs since no electron transfer processes occur in Au/PSS and Au/SiO2core/shell nanocomposites.
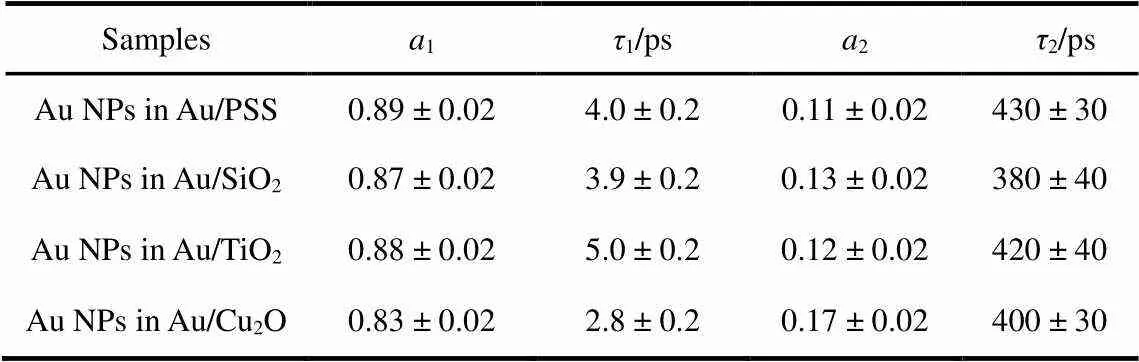
Table 1 Fitting parameters for the plasmon bleaching recovery kinetics of Au NPs in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O core/shell nanocomposites.
A summation of two exponential decay functions was used.
3.4 Interband transition decay kinetics of Au NPs in Au/TiO2 and Au/Cu2O core/shell nanocomposites
To confirm the time constant1listed in Table 1 for Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites are the electron recombination time from the conduction band of TiO2and Cu2O to the Fermi level of Au NPs, we further recorded their respective interband transition decay kinetics, as shown in Fig.4.
Both the interband transition decay of Au NPs in Au/TiO2core/shell nanocomposite and the interband transition decay of Au NPs in Au/Cu2O core/shell nanocomposite need a summation of two exponential decay functions to be well fitted. The fit for the interband transition decay of Au NPs in Au/TiO2core/shell nanocomposite gave two time constants (amplitudes) of 4.9 ± 0.2 (0.92 ± 0.02) and 360 ± 50 ps (0.08 ± 0.02). And the fit for the interband transition decay of Au NPs in Au/Cu2O core/shell nanocomposite gave two time constants (amplitudes) of 2.8 ± 0.2 (0.93 ± 0.02) and 390 ± 60 ps (0.07 ± 0.02). The interband transition decays gave the almost identical time constants as the plasmon bleaching recoveries did, which is further suggested that the obtained shorter time constants from the plasmon bleaching recovery kinetics of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites are the electron recombination time from the conduction band of TiO2and Cu2O to the Fermi level of Au NPs. The result that the electron recombination rate from the conduction band of TiO2to the Fermi level of Au NPs in Au/TiO2core/shell nanocomposite is smaller than the electron recombination rate from the conduction band of Cu2O to the Fermi level of Au NPs in Au/Cu2O core/shell nanocomposite is consistent with the experimental observation which the energy level of the conduction band of TiO2is lower than energy level of the conduction band of Cu2O. In addition, the obtained almost identical time constants from both the plasmon bleaching recovery and the interband transition decay of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites also implied that the electron transfer from the excited plasmon band of Au NPs to the conduction band of TiO2and Cu2O are very fast (within our time resolution, ~200 fs), which is consistent with the previous studies27.
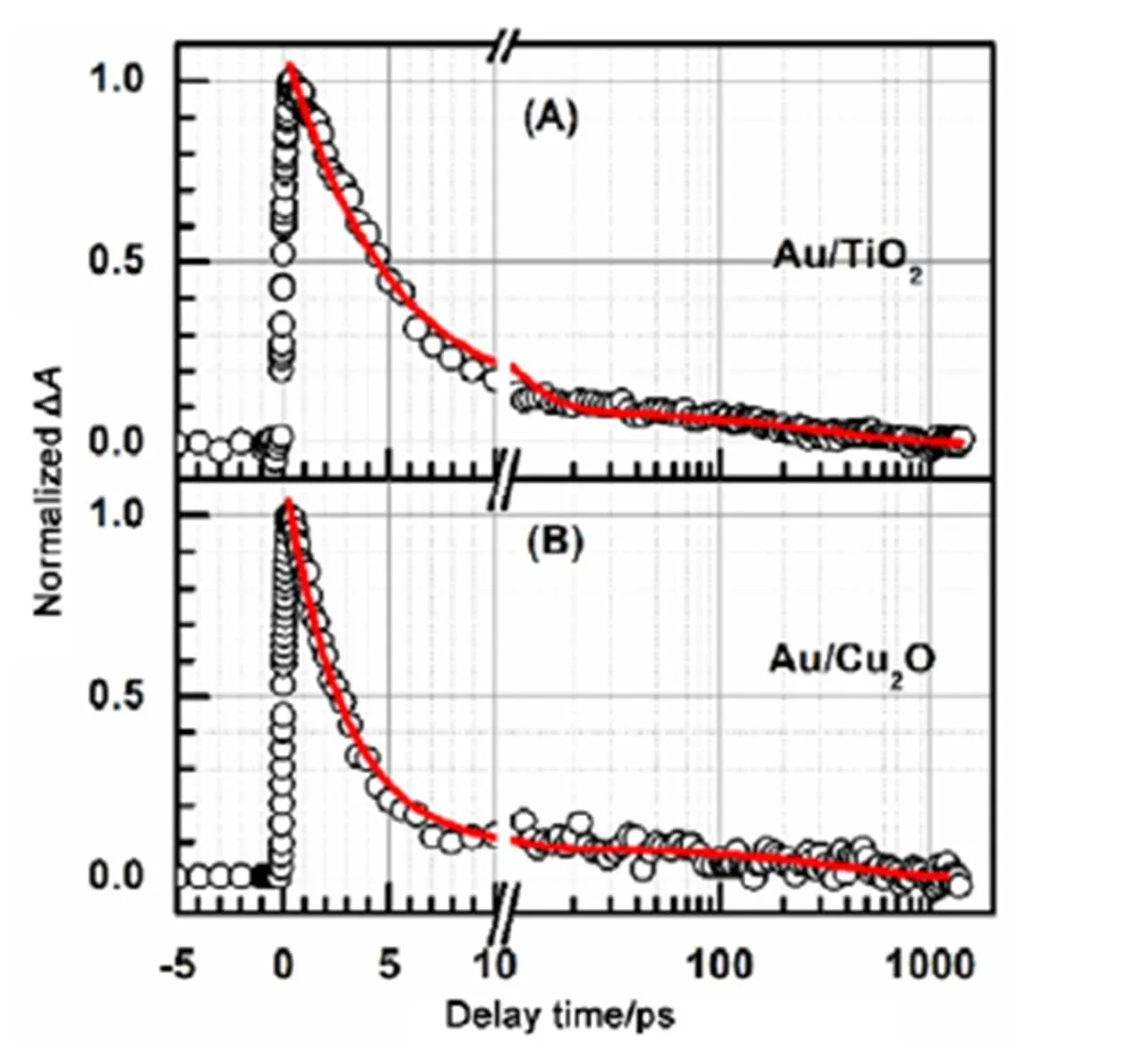
Fig.4 Interband transition decay kinetics of Au NPs in Au/TiO2 (A) and Au/Cu2O (B) core/shell nanocomposites.
The red lines are the fits. The pump/probe wavelengths for Au NPs in Au/TiO2and Au/Cu2O nanocomposites are 525/485, and 545/500 nm, respectively.
4 Conclusions
In summary, we investigated the interaction between Au NPs and its surrounding medium in Au/PSS, Au/SiO2, Au/TiO2, and Au/Cu2O core/shell nanocomposites by using both steady-state absorption and femtosecond transient absorption measurements. We found that the plasmon absorption band of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites are much broader than those in Au/PSS and Au/SiO2core/shell nanocomposites. We also found that the intensity ratio between the positive absorption band at the blue side of the plasmon bleaching band and the positive absorption band at the red side of the plasmon bleaching band in the femtosecond transient absorption spectra of Au NPs in Au/TiO2and Au/Cu2O core/shell nanocomposites are dramatically larger than those in Au/PSS and Au/SiO2core/shell nanocomposites. More importantly, we found that the excited electron of Au NPs after an excitation on its resonance plasmon band can be transferred into the conduction band of TiO2and Cu2O in Au/TiO2and Au/Cu2O core/shell nanocomposites.
Acknowledgement: The authors appreciate Dr. Weiwei He from Xuchang University for his help on the sample preparation.
Supporting information: available free of chargethe internet at http://www.whxb.pku.edu.cn.
(1) Daniel, M. C.; Astruc, D.2004,, 293. doi: 10.1021/cr030698+
(2) El-Sayed, I. H.; Huang, X.; El-Sayed, M. A.2006,, 129. doi: 10.1016/j.canlet.2005.07.035
(3) El-Sayed, I. H.; Huang, X. H.; El-Sayed, M. A.2005,, 829. doi: 10.1021/nl050074e
(4) Kamat, P. V.2002,, 7729. doi: 10.1021/jp0209289
(5) Kamat, P. V.2007,, 2834. doi: 10.1021/jp066952u
(6) Maeda, K.; Domen, K.2010,, 2655. doi: 10.1021/jz1007966
(7) Sperling, R. A.; Rivera Gil, P.; Zhang, F.; Zanella, M.; Parak, W. J.2008,, 1896. doi: 10.1039/b712170a
(8) Hartland, G. V.2011,, 3858. doi: 10.1021/cr1002547
(9) Link, S.; El-Sayed, M. A.2003,, 331. doi: 10.1146/annurev.physchem.54.011002.103759
(10) Link, S.; El-Sayed, M. A.2000,, 409. doi: 10.1080/01442350050034180
(11) Smith, B. A.; Zhang, J. Z.; Giebel, U.; Schmid, G.1997,, 139. doi: 10.1016/S0009-2614(97)00339-4
(12) Perner, M.; Bost, P.; Lemmer, U.; vonPlessen, G.; Feldmann, J.; Becker, U.; Mennig, M.; Schmitt, M.; Schmidt, H.1997,, 2192. doi: 10.1103/PhysRevLett.78.2192
(13) Inouye, H.; Tanaka, K.; Tanahashi, I.; Hirao, K.1998,, 11334. doi: 10.1103/PhysRevB.57.11334
(14) Link, S.; Furube, A.; Mohamed, M. B.; Asahi, T.; Masuhara, H.; El-Sayed, M. A.2002,, 945. doi: 10.1021/jp013311k
(15) Ahmadi, T. S.; Logunov, S. L.; El-Sayed, M. A.1996,, 8053. doi: 10.1021/jp960484e
(16) Hartland, G. V.2004,, 5263. doi: 10.1039/B413368D
(17) Logunov, S. L.; Ahmadi, T. S.; El-Sayed, M. A.; Khoury, J. T.; Whetten, R. L.1997,, 3713. doi: 10.1021/jp962923f
(18) Hodak, J. H.; Henglein, A.; Hartland, G. V.1999,, 8613. doi: 10.1063/1.480202
(19) Hodak, J. H.; Martini, I.; Hartland, G. V.1998,, 6958.doi: 10.1021/jp9809787
(20) Link, S.; El-Sayed, M. A.1999,, 4212. doi: 10.1021/jp984796o
(21) Mohamed, M. B.; Ahmadi, T. S.; Link, S.; Braun, M.; El-Sayed, M. A.2001,, 55. doi: 10.1016/S0009-2614(01)00653-4
(22) Subramanian, V.; Wolf, E. E.; Kamat, P. V.2004,, 4943. doi: 10.1021/ja0315199
(23) Tian, Y.; Tatsuma, T.2005,, 7632. doi: 10.1021/ja042192u
(24) Chen, M.; Goodman, D. W.2008,, 1860. doi: 10.1039/b707318f
(25) Comotti, M.; Li, W. C.; Spliethoff, B.; Schuth, F.2006,, 917. doi: 10.1021/ja0561441
(26) Du, L.; Furube, A.; Hara, K.; Katoh, R.; Tachiya, M.2013,, 21. doi: 10.1016/j.jphotochemrev.2012.11.001
(27) Furube, A.; Du, L.; Hara, K.; Katoh, R.; Tachiya, M.2007,, 14852. doi: 10.1021/ja076134v
(28) Wu, K.; Rodriguez-Cordoba, W. E.; Yang, Y.; Lian, T.2013,, 5255. doi: 10.1021/nl402730m
(29) Zhang, Y.; Yuan, S.; Lu, R.; Yu, A.2013,, 7308. doi: 10.1021/jp404466f
(30) Wang, J.-D.; Li, S.; Lu, R.; Yu, A.-C.2015,, 1787. [王经东, 李 爽, 吕 荣, 于安池. 物理化学学报, 2015,, 1787.] doi: 10.3866/PKU.WHXB201507241
(31) Johnson, P. B.; Christy, R. W.1972,, 4370. doi: 10.1103/PhysRevB.6.4370
(32) Aiboushev, A.; Gostev, F.; Shelaev, I.; Kostrov, A.; Kanaev, A.; Museur, L.; Traore, M.; Sarkisov, O.; Nadtochenko, V.2013,, 631. doi: 10.1039/c2pp25227a
(33) Karam, T. E.; Khoury, R. A.; Haber, L. H.2016,, 124704. doi: 10.1063/1.4944385
金纳米颗粒在不同包裹介质中的超快等离子体动力学
陈晓宇 王经东 于安池*
(中国人民大学化学系,北京 100872)
为了探究不同包裹介质对金纳米颗粒的超快等离子体动力学的影响,本文我们制备了聚苯乙烯磺酸钠(PSS)、二氧化硅(SiO2)、二氧化钛(TiO2)以及氧化亚铜(Cu2O)四种介质包裹的金纳米颗粒。运用飞秒泵浦探测技术,我们分别获得了PSS、SiO2、TiO2以及Cu2O包裹的金纳米颗粒在不同时间延迟下的飞秒瞬态吸收光谱。我们发现TiO2包裹的金纳米颗粒的飞秒瞬态吸收光谱特征与PSS和SiO2包裹的金纳米颗粒的飞秒瞬态吸收光谱特征有很大的不同,但与Cu2O包裹的金纳米颗粒的飞秒瞬态吸收光谱特征相似。全面分析PSS、SiO2、TiO2和Cu2O包裹的金纳米颗粒的超快等离子体动力学行为,我们发现在TiO2和Cu2O包裹的金纳米颗粒体系中光激发金纳米颗粒的等离子体共振吸收后产生的激发电子可以有效地转移到TiO2和Cu2O的导带上。
飞秒瞬态吸收光谱;等离子体共振激发;电子转移;金纳米颗粒;核/壳结构
O643
10.3866/PKU.WHXB201705222
April 26, 2017;
May 15, 2017;
May 22, 2017.
Corresponding author. Email: a.yu@chem.ruc.edu.cn; Tel: +86-10-6251-4601.
The project was supported by the National Natural Science Foundation of China (21373269).
国家自然科学基金(21373269)资助项目
猜你喜欢
人人健康(2021年16期)2021-12-01
防爆电机(2020年3期)2020-11-06
制造技术与机床(2019年8期)2019-09-03
华东师范大学学报(自然科学版)(2019年3期)2019-06-24
中国资源综合利用(2017年1期)2018-01-22
山东工业技术(2016年15期)2016-12-01
系统工程与电子技术(2016年2期)2016-04-16
中国粮油学报(2016年5期)2016-01-23
中国光学(2015年5期)2015-12-09
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
