
溶剂热法一锅合成亚甲基二芳胺、三亚甲基三芳胺和四亚甲基四芳胺类化合物
2017-12-26 05:36陈连清杜艳婷郭志伟
中南民族大学学报(自然科学版) 2017年4期
陈连清,杜艳婷,周 泉,郭志伟
(中南民族大学 化学与材料科学学院 催化材料科学国家民委-教育部共建重点实验室, 武汉 430074)
溶剂热法一锅合成亚甲基二芳胺、三亚甲基三芳胺和四亚甲基四芳胺类化合物
陈连清,杜艳婷,周 泉,郭志伟
(中南民族大学 化学与材料科学学院 催化材料科学国家民委-教育部共建重点实验室, 武汉 430074)
为寻找一种更高效简便的合成亚甲基芳胺类化合物的方法,以芳胺和多聚甲醛为原料,在不同条件和溶剂中使用溶剂热法一锅合成了一系列的亚甲基二芳胺、三亚甲基三芳胺和四亚甲基四芳胺类化合物,并通过红外光谱、核磁共振、元素分析等手段进行了结构表征,考察了苯环上不同取代基及反映时间对产物的影响.结果表明:溶剂热法具有后处理简便、产率提高的优势,是一种合成亚甲基二芳胺、三胺、四胺类衍生物的新的通用方法,这些化合物具有良好的荧光性能.
芳香胺;多聚甲醛;溶剂热法;亚甲基二芳胺;三亚甲基三芳胺;四亚甲基四芳胺
亚甲基芳胺类化合物在化工、医药、工业中有重要作用,其中亚甲基二芳胺衍生物可作为参与构建金属有机化合物的配体[1]和用于研究Mannich反应机理的Mannich试剂[2].三亚甲基三芳胺即氢化三嗪类物质如黑索金、化学防治剂DHT(2,4-二氧六氢-1,3,5三嗪)等,被广泛应用于化工、医药、生物等领域,亚甲基四亚芳胺类化合物如二硝基五亚甲基四胺(又称3,7-二硝基-1,3,5,7-四氮杂双环[3.3.1]壬烷),俗称HMT,是综合性能最优良的单质炸药之一[3].
亚甲基二芳胺衍生物可由相应的伯胺与二碘甲烷或二溴甲烷在碱性条件下制备[4].吕银祥等[1]使用N,N-二甲基甲酰胺(DMF)与仲胺为原料制备亚甲基二芳胺衍生物,对反应条件要求较为苛刻,需要在氮气的保护下进行.氢化三嗪可微波制备[5],但大部分使用常规加热回流法制备.也有以发烟硝酸、尿素、甲醛和氨水为原料一锅合成二硝基五亚甲基四芳胺,但所用的原料具有较强的氧化性,实验后续处理较为繁杂.以上方法不是由相同的原料一步合成出三类亚甲基芳胺类化合物,不利于实际生产.所以迫切需要一种原料简单易得,操作简便,产率较高的通用合成方法.原料同为芳胺和甲醛的情况下,Giumanini等[6]在惰性溶剂中加碳酸钠为催化剂制备亚甲基二芳胺;Singh[7]用37%的乙醇溶液为溶剂合成三亚甲基三胺类化合物.Ghandi[8]在甲酸存在下,使用N,N′-二芳基亚甲基二芳胺在乙腈中与甲醛水溶液制备了四亚甲基四芳胺化合物.也就是说,在不同的溶剂下,可通过调整摩尔比和反应条件等来合成不同的产物.
由于溶剂热法将原料和有机溶剂置于高温高压密闭容器中反应,不仅可有效防止有毒物质的挥发,还使反应物的化学反应活性大大提高[9,10].因此本文以芳胺和甲醛为原料,在不同条件下用溶剂热法一锅合成亚甲基二芳胺、三亚甲基三芳胺、四亚甲基四芳胺类化合物,实验证实它们具有稳定的光物理性能和良好的荧光性能.由于亚甲基芳胺在医药、生物上的运用,其良好的荧光性可为其作为药物残余检测、生物体内荧光探针等功能开发提供支持.
1 实验部分
1.1 试剂和仪器
苯胺、对甲基苯胺、对氟苯胺、氟虫腈、对氨基苯甲酸乙酯、对氨基苯腈、N,N-二甲基对苯二胺、甲苯、乙酸乙酯、硅胶粉、乙醇等均为市售分析纯.
恒温加热磁力搅拌器(DF-101S,上海东玺制冷仪器),低温冷却液循环泵(DLSB-5L/20,巩义市予华仪器),旋转蒸发器(RE-52,上海亚荣生化仪器厂),循环水真空泵[SHZ-D(III)型,武汉科尔];三用紫外线分析仪(ZF-1型,上海嘉鹏),电磁式真空泵(ACO系列,森森集团),数字熔点仪(WRS-1B,上海精科),傅立叶红外光谱仪(NEXUS470型,美国尼高力),全数字化核磁共振谱仪(AVANCE III 400 MHz,瑞士布鲁克),紫外-可见分光光度计(Lambda-Bio35,日本岛津),荧光光谱仪(PE LS-55 型,美国),元素分析仪(Vario-EL III CHNS型,美国).
1.2 合成方法
1.2.1 加热回流法
取干燥的100 mL烧杯,称量0.02 mol芳胺,再倒入30 mL甲苯,溶解后转移至干燥的100 mL的单口烧瓶,并将0.02 mol多聚甲醛(0.6 g)加入其中,在烧瓶上端加入冷凝管并用干燥管封口,防止溶剂挥发,在磁力搅拌器中搅拌加热回流2 h.反应结束后趁热过滤,使用硅胶吸附并干法制样,用洗脱剂体积比为1︰10的乙酸乙酯与石油醚经柱层析法提纯,得到产物.
1.2.2 溶剂热法
在100 mL的烧杯中加入0.02 mol芳胺,以及0.02 mol(0.6 g)多聚甲醛,加甲苯 30 mL,转移至干燥100 mL聚四氟乙烯内衬中,将聚四氟乙烯内衬装入高压反应釜中,放入烘箱中在120℃下加热2 h.反应结束后冷却至室温,使用旋蒸仪把甲苯旋干.加入少量乙酸乙酯溶解,再加少量硅胶粉,之后旋干至粉末.用洗脱剂体积比为1︰10的乙酸乙酯与石油醚经柱层析法提纯,得到产物(合成路线见图1).
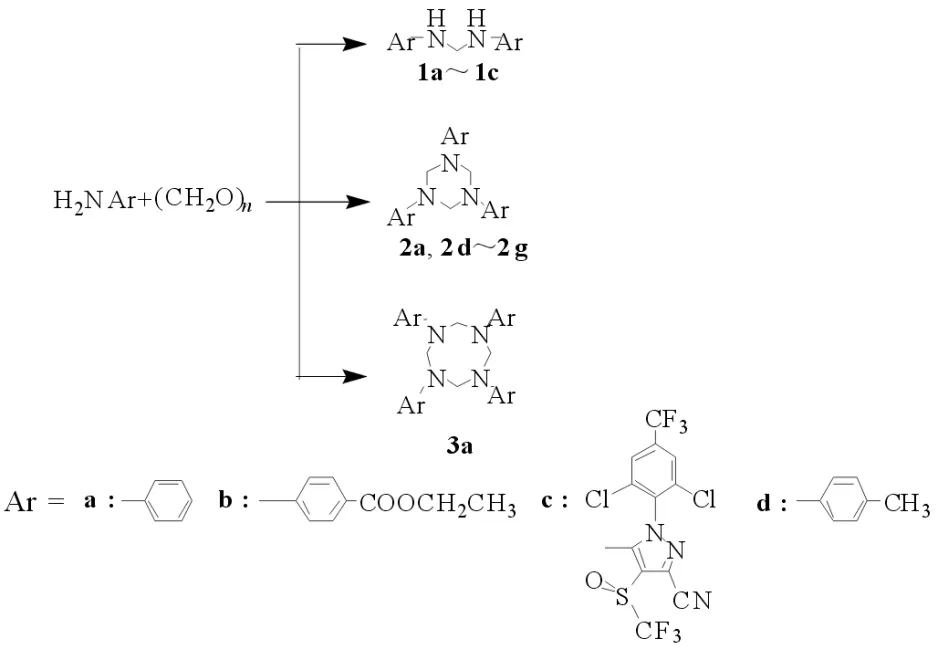
图1 亚甲基二芳胺、三亚甲基三芳胺、四亚甲基四芳胺的合成路线Fig.1 Synthetic route of methylene diarylamine, trimethylene triarylamine, tetramethylene tetraarylamine
2 实验结果
2.1 亚甲基二芳胺类物质的合成
亚甲基二芳胺1a~1c的合成路线见图2.其中产物1a:收率94%[8].m.p.61℃1H NMR(CDCl3, DMSO, 400 MHz)δ: 4.47 (t,2 H), 6.14(t, 2 H), 6.38~7.25(m, 10 H ); IR (KBr,ν/cm-1): 3030, 3421, 3374, 1515, 1503, 869.Anal.calc for C13H14N2:C 78.75, H 7.12, N 14.13;found: C 78.64, H 7.23, N 14.16.MS (FAB):m/e, 198 (M+).
产物1b:收率34.8%.m.p.195.4~195.8℃1H NMR(CDCl3, DMSO, 400 MHz)δ:7.69~7.71(d,4 H), 7.25~7.28(t, 2 H), 6.72~6.74(d, 4 H), 4.56~4.59(t,2 H),4.18~4.23(q,4 H),1.25~1.29(t,6 H);IR (KBr,ν/cm-1): 3368,2987,1685,1599,1531,1373,1171,1025,843.Anal.calc for C19H2N2O4: C 66.65,H 6.48,N 8.18,O 18.69;found: C 66.73,H 6.42,N 8.21,O 18.69.MS (FAB):m/e,342 (M+).
产物1c:收率18.3%.m.p.162.2~165.2℃1H NMR (CDCl3,DMSO,400 MHz)δ: 7.90(s,2 H),7.85(s,2 H),7.02(t,2 H),4.85(t,2 H);IR (KBr,ν/cm-1): 3291,3087,2246,1689,1556,1389,1313,1184,819.Anal.calc for C25H8Cl4F12N8O2S2: C 33.88,H 0.91,Cl 16.00,F 25.72,N 12.64,O 3.61,S 7.24;found: C 33.67,H 0.98,N 12.54,Cl 16.11,F 25.59,O 3.74,S 7.36.MS (FAB):m/e,883 (M+).
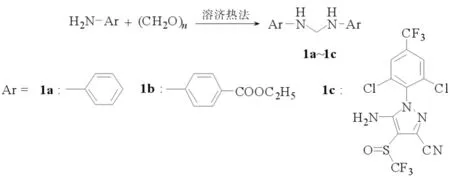
图2 亚甲基二芳胺 1a~1c 的合成路线Fig.2 Synthesis route of methylene diarylamine 1a-1c
2.2 三亚甲基三芳胺(氢化三嗪)类物质的合成
三亚甲基三芳胺2a,2b~2h的合成路线见图3.产物2a:收率41.6%.m.p.145.6~147.1℃1H NMR(CDCl3,DMSO,400 MHz)δ: 7.21~7.26(s,6H),7.02~7.04 (s,6H),6.86~6.90(s,3H),4.9(s,6H); IR (KBr,ν/cm-1): 3030,2887,1500,1372,1164,749.Anal.calc for C12H12N3: C 79.97,H 6.71,N 13.32;found: C 50.70,H 2.18,N 13.15.MS (FAB):m/e,315 (M+).
产物2d:收率58.1%.m.p.162.2~165.2℃1H NMR(CDCl3,DMSO,400 MHz)δ: 6.95(s,12 H),4.74(s,6H),2.17(s,9H); IR (KBr,ν/cm-1): 3026,2860,1516,1326. Anal.calc for C24H27N3: C 80.63,H 7.61,N 11.75;found: C 80.73,H 7.92,N 12.11.MS (FAB):m/e,357 (M+).
产物2e:收率52.8%.m.p.160℃1H NMR(CDCl3,DMSO,400 MHz)δ: 6.88~7.00(m,12 H),4.77(s,6H); IR (KBr,ν/cm-1): 3011,1510,1229,1163,820.Anal.calc for C21H18F3N3: C 68.28,H 4.91,F 15.43,N 11.38;found:C 68.28,H 4.91,F 15.43,N 11.38.MS (FAB):m/e,369(M+).
产物2f:收率91.7%.m.p.245℃1H NMR(CDCl3,DMSO,400 MHz)δ:5.20(s,6H),7.39(q,12H); IR (KBr,ν/cm-1): 3096,2220,1393,1367,1239,830.Anal.calc for C24H18N6: C 73.8,H 4.65,N 21.52;found: C 73.8,H 4.65,N 21.52.MS (FAB):m/e,390 (M+).
产物2g:收率90.0%.m.p.158℃1H NMR(CDCl3,DMSO,400 MHz)δ: 2.83(s,6H),4.58(s,6H),6.84(q,12H); IR (KBr,ν/cm-1): 3075,2875,1510,1477,1388,1227,815[9].Anal.calc for C27H36N6: C 72.94,H 8.16,N 18.90;found: C 72.94,H 8.16,N 18.90.MS (FAB):m/e,444 (M+).
产物2h:收率96.0%.m.p.131℃1H NMR(CDCl3,DMSO,400 MHz)δ: 3.73(s,3H),4.69(s,6H),6.95(q,12H); IR (KBr,ν/cm-1): 3065,3040,2840,1505,1380,1295,1033,828.Anal.calc for C24H27N3O3: C 71.09,H 6.71,N 10.36,O 11.84;found: C 71.09,H 6.71,N 10.36,O 11.84.MS (FAB):m/e,405 (M+).
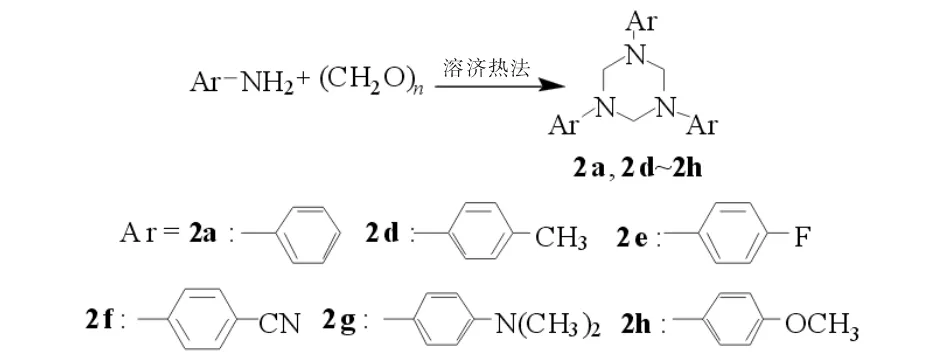
图3 三亚甲基三芳胺 2a,2b~2h 的合成路线Fig.3 Synthesis route of trimethylene triarylamine 2a,2b-2h
2.3 四亚甲基四芳胺的合成
四亚甲基四芳胺3a的合成路线见图4.产物3a:收率20.0%.m.p.304℃1H NMR(CDCl3,DMSO,400 MHz)δ: 4.90(s,8 H),6.70~7.54 (m,20 H),IR (KBr,ν/cm-1): 3030,2883,1515,1322,1128,743.Anal.calc for C12H7N3O2: C 79.97,H 6.71,N 13.32;found C 79.97,H 6.71,N 13.32.MS (FAB):m/e,420(M+).
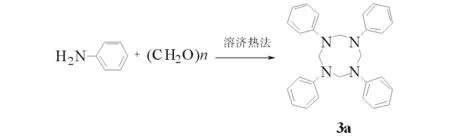
图4 四亚甲基四芳胺 3a 的合成路线Fig.4 Synthesis route of tetramethylene tetraarylamine 3a
3 分析与讨论
3.1 产物结构表征
化合物1a~3a的物性常数及元素分析组成数据见表1.由表1可见:目标化合物的C、H测定值与理论组成一致.
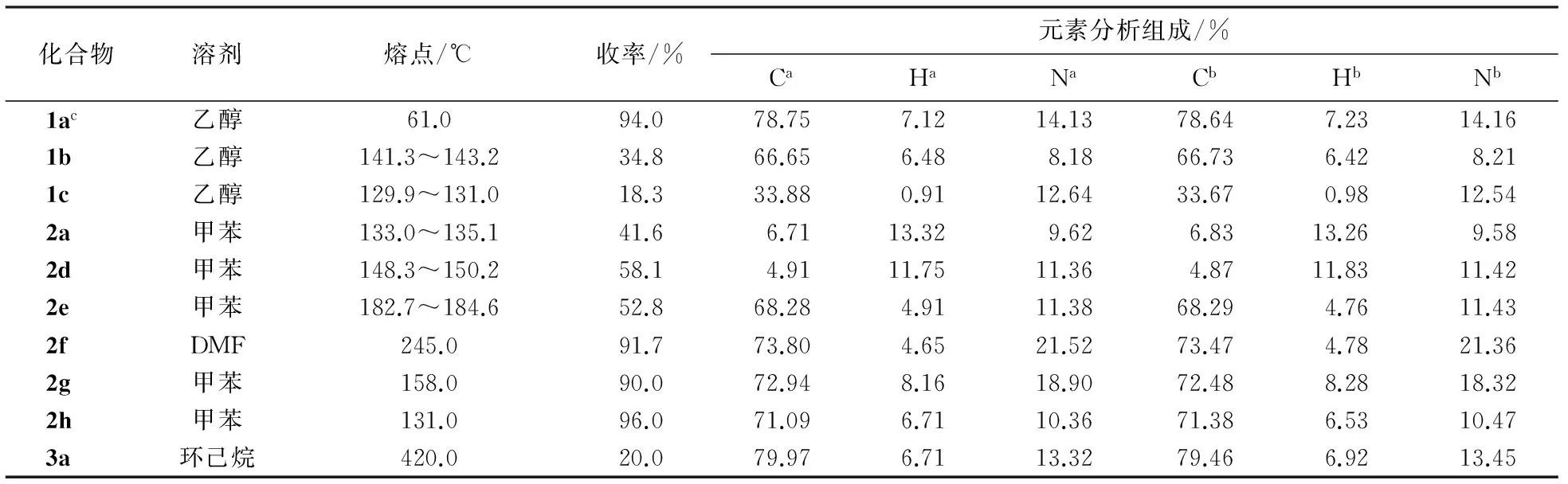
表1 化合物1a~3a的物性常数及元素分析组成
注:a为理论值;b为实验值; c为参考文献值
化合物的红外和质谱数据见表2.由表2可见:目标化合物各个特征吸收振动峰均得到证实,如苯环的特征吸收峰1a~1c分别为3030,2987,3087 cm-1,2a、2d、2e分别为3030,3026,3011 cm-1等;胺的N―H特征吸收峰,1a~1c分别为3421,3368,3291 cm-1,而三亚甲基三芳胺和四亚甲基四芳胺产物由于氮上没有氢,因此2a、2d、2e等化合物没有N―H吸收峰.亚甲基芳胺C―N吸收峰,2a、2d、2e分别为1164,1240,1229 cm-1等,均有所归属.在MS谱图中目标化合物的分子碎片峰符合分子理论组成.
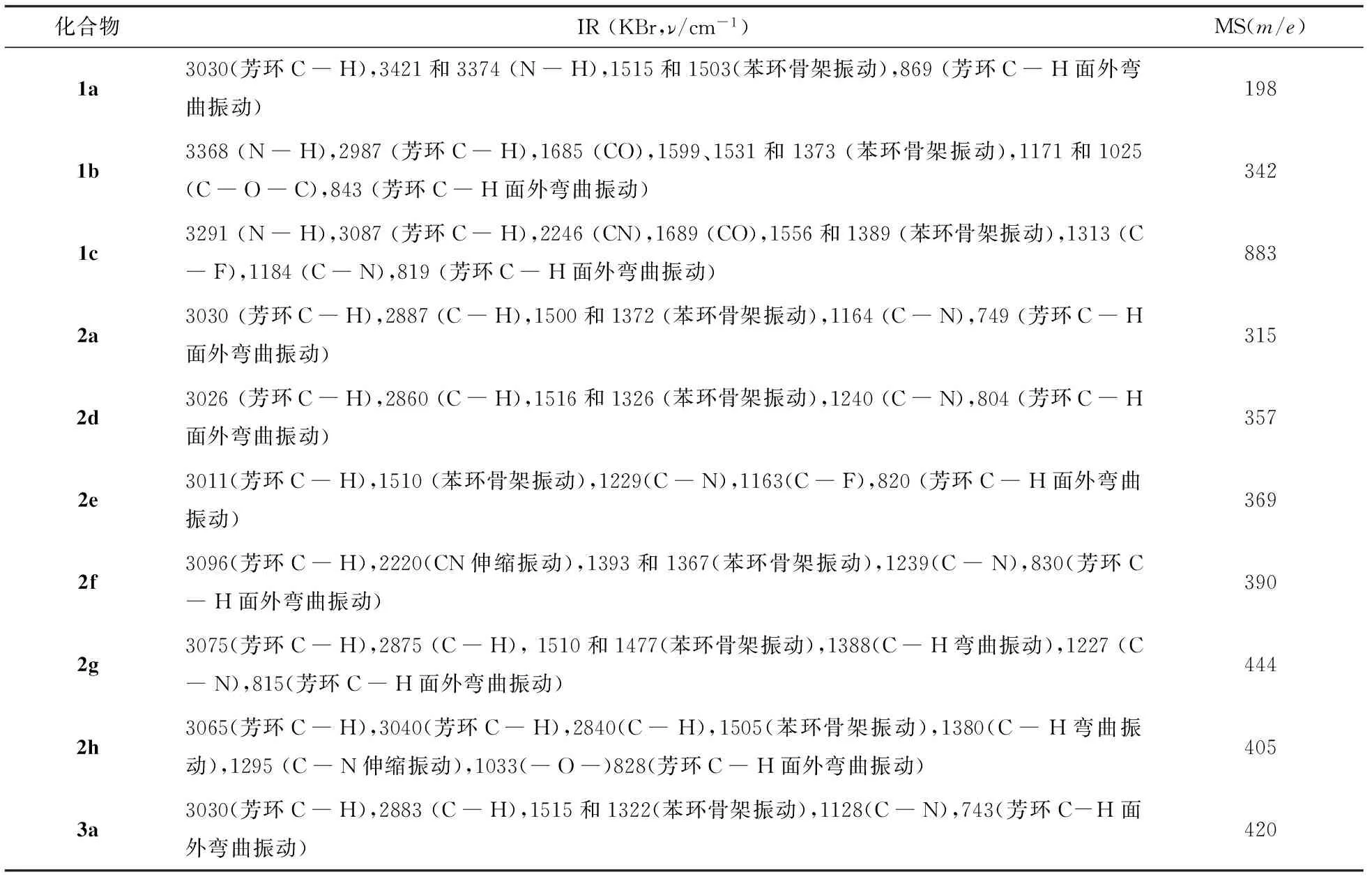
表2 化合物的红外和质谱数据Tab.2 IR and MS data of compounds
化合物1a~3a的1H NMR谱数据见表3.由表3可见:苯环上的H峰如1c为7.90(s,2H,Ar―H),氨基上的H峰如1c7.02(t,2H,N―H)以及亚甲基上的H峰如2e上的4.77(s,6H,CH―H);各种质子的化学位移均有所归属.以上表征数据说明目标化合物的结构得到确认.

表3 化合物1a~3a1H NMR数据Tab.3 1H NMR data of compound 1a-3a
3.2 反应机理推测
在芳胺和甲醛的的反应过程中,推测其反应机理(见图5),甲醛与芳胺反应生成中间体亚甲基二芳胺[11],亚甲基二芳胺继续反应生成二亚甲基三芳胺后再与一分子甲醛发生醛胺缩合合环得到三亚甲基三芳胺(氢化均三嗪);其中二亚甲基三芳胺也可继续反应生成三亚甲基四芳胺,三亚甲基四芳胺再与一分子甲醛反应合环得到四亚甲基四芳胺.要想产物停留在二胺阶段,可通过改变胺和甲醛的摩尔比达到目的.但是醛胺合环时,若胺所连接的基团较大会产生较大的空间位阻,不利于合环.因此,产物1b和1c停留在亚甲基二芳胺阶段,是由于空间位阻导致.
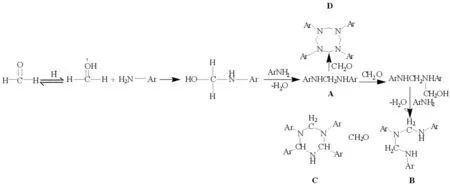
图5 反应机理推测Fig.5 Speculation of reaction mechanism
Ghandi用摩尔比为3︰2的2-氨基嘧啶与甲醛反应,在反应过程中通过薄层色谱(TLC)检测到了二亚甲基三芳胺类产物B的存在,产物B再与额外的甲醛反应生成了产物C[8].Randaccio[12]报道了芳胺和多聚甲醛在甲苯中的回流条件反应合成了四亚甲基四芳胺,推测了亚甲基二芳胺为中间体,为证明此结论,本文研究了反应时间对亚甲基二芳胺产率的影响,结果见图6.由图6可见:亚甲基二芳胺化合物1b产率随着反应时间的增加而加大,说明亚甲基二芳胺存在并为中间体,而产物1b不能进一步反应形成三亚甲基三芳胺和四亚甲基四芳胺产物,可能与其空间位阻有关.Giumanini等[7]合成了大量的亚甲基亚芳胺类产物,其中包括1a、2f、2g、2h、3a,通过核磁红外等手段证明了各类亚甲基亚芳胺产物的存在.
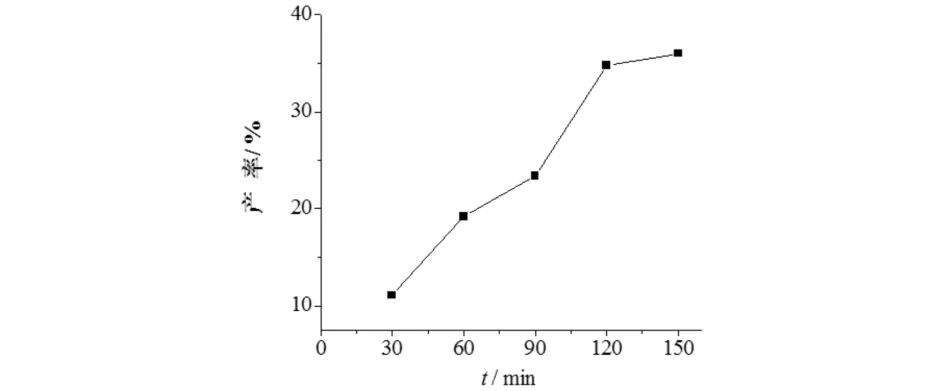
图6 反应时间对亚甲基二芳胺产率的影响Fig.6 Effect of reaction time on the yield of methylene diarylamine
3.3 合成方法比较
已有研究以苯二胺为原料,通过超声波辅助,使用芳胺和甲醛水溶液在乙醇中加热回流反应,或者以氟苯胺和水甲醛为原料,微波法合成三亚甲基三芳胺;这两种反应后续处理包括静置沉淀、过滤、萃取,旋蒸、柱层析.而溶剂热法较常规的加热回流法和超声法更有效率.本文以4种产物为例,加热时间均为2 h,发现溶剂热法产率较常规加热高(见表4).
表4 不同合成方法和反应时间的产率对比表
Tab.4 Yield comparison of different synthesis methods and different reaction time
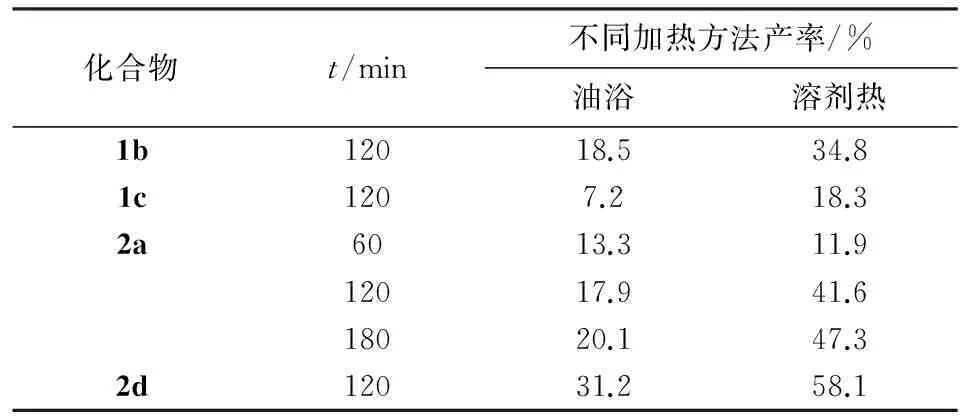
化合物t/min不同加热方法产率/%油浴溶剂热1b12018.534.81c1207.218.32a6013.311.912017.941.618020.147.32d12031.258.1
注:化合物1b和1c的加热溶剂为乙醇,化合物2a和2d的加热溶剂为甲苯
化合物2a在 1 h时溶剂热法产率低于常规油浴加热,由于加热时间太短而原料尚未完全溶解,此时回流加热更能充分反应.加热3 h,在原料完全溶解情况下,溶剂热法产率较常规加热回流法相比大大提高.且溶剂热法反应后无原料残留,后续处理更简单;反应釜冷却后有晶体析出,证明反应更完全.它还能灵活多变地与其他反应方法结合.因此,溶剂热法是一种以芳胺和多聚甲醛为原料,在不同条件下制备亚甲基二芳胺、三亚甲基三芳胺、四亚甲基四芳胺类化合物通用的方法.
3.4 产物的光物理性能
表5和图7为相同条件下化合物1b、1c、2a、2d、2e的紫外吸收光谱,由表5和图7可见:这类化合物的紫外吸收光谱主要集中在苯环的B带吸收,化合物1b在272.50 nm有强吸收峰,在227.50 nm有宽带吸收峰;化合物1c在296.50 nm有强吸收峰并发生了红移,在226.00 nm有宽带吸收峰;化合物2a和2d均在251.50 nm有强吸收峰;化合物2e在288.00 nm有强吸收峰,在243.00 nm有宽峰并出现红移.

表5 化合物1b、1c、2a、2d、2e紫外吸收光谱数据Tab.5 UV absorption spectrum data of compound 1b,1c,2a,2d,2e
注:a表示紫外溶剂为CH2Cl2; b表示固体荧光; c表示荧光强度
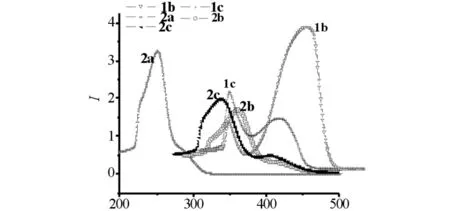
λ/nm图7 化合物 1b、1c、2a、2d、2e 紫外光谱Fig.7 UV spectra of compound 1b,1c,2a,2d and 2e
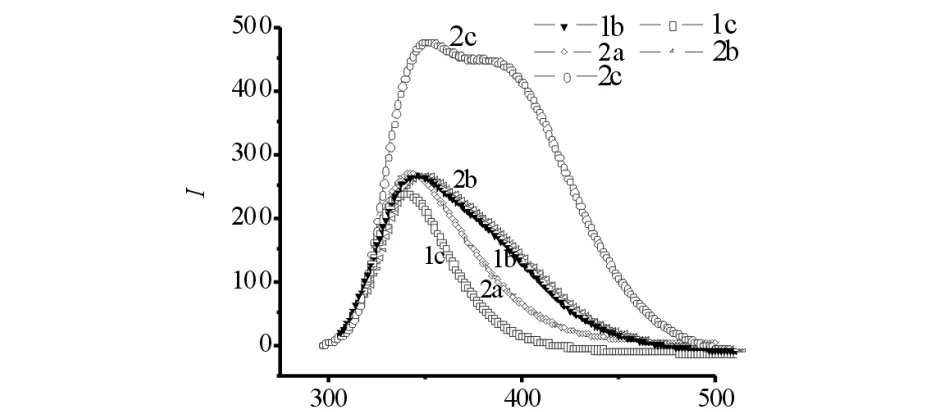
λ/nm图8 化合物 1b、1c、2a、2d、2e 的荧光光谱 Fig.8 Fluorescence spectra of compound 1b,1c,2a,2d and 2e
化合物1b、1c、2a、2d、2e的荧光光谱见图8.由图8可见:它们具有的荧光发射峰在320~350 nm,1b、1c为亚甲基二芳胺产物,其荧光发射峰比亚甲基三芳胺小,但又保持在一定的范围内,说明这一类化合物的荧光发射主要集中在苯环上,苯环间由不同数目的碳氮键连接,荧光发射峰增大.亚甲基二芳胺产物只有2个苯环,三亚甲基三芳胺有3个苯环,所以荧光强度增强.三亚甲基三芳胺上连接不同的取代基也是荧光发射峰和荧光强度发生改变的原因,但是它们的荧光发射峰的变化范围不是很大,说明其光物理性能稳定,荧光性能良好,使之成为生物荧光探针及为亚甲基亚芳胺类物质成分残留检测提供了支持.
3.5 晶体结构
化合物1c的分子结构图和结构堆积图分别见图9和图10,它的分子通过碳氮键连接两个苯基吡唑骨架组成.
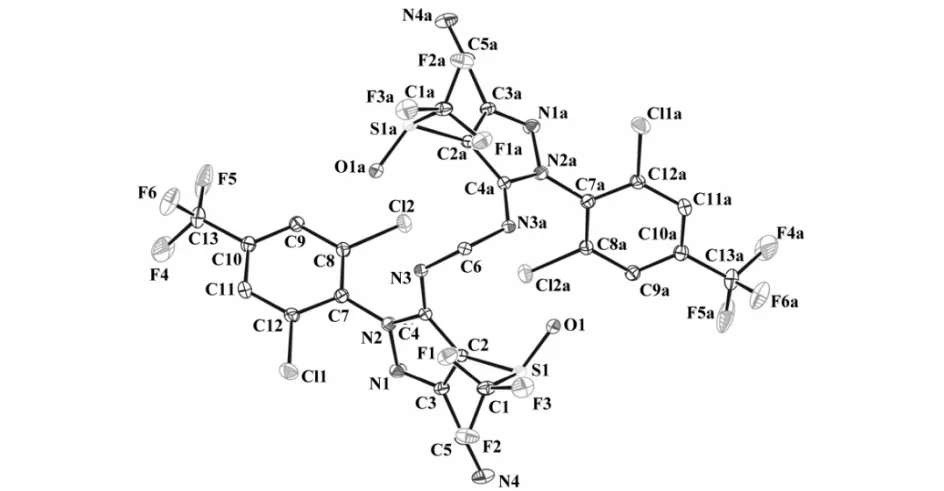
图9 化合物 1c 的分子结构图Fig.9 Molecular structure of compound 1c
化合物1c的晶胞参数、部分键长数据和部分键角数据分别见表6、表7和表8.由表6~8可知:晶胞参数a=20.0584(9) Å,b=16.1373(7) Å,c=10.6232(5) Å,α=90°,β=93.283°,γ=90°晶胞的体积为3433.1(3) Å3,苯环内键角的理想角度为120°[13,14],而实际角度
介于118~120°.N(3)―C(6)的键长为1.342(6) Å,表示碳氮单键;C(8)―C(9)―H(9)的键角为116.8(13)°,N(2)―N(1)―C(2)的键角为118.5(4)°,属于正常的键角范围之内.产物1c中吡唑环的苯环和五元环以一定的扭曲角存在,两个吡唑环骨架由碳氮键连接起来的,两个骨架之间的连接处呈C(4)―N(3)―C(6)成133.6(5)°的二面角.
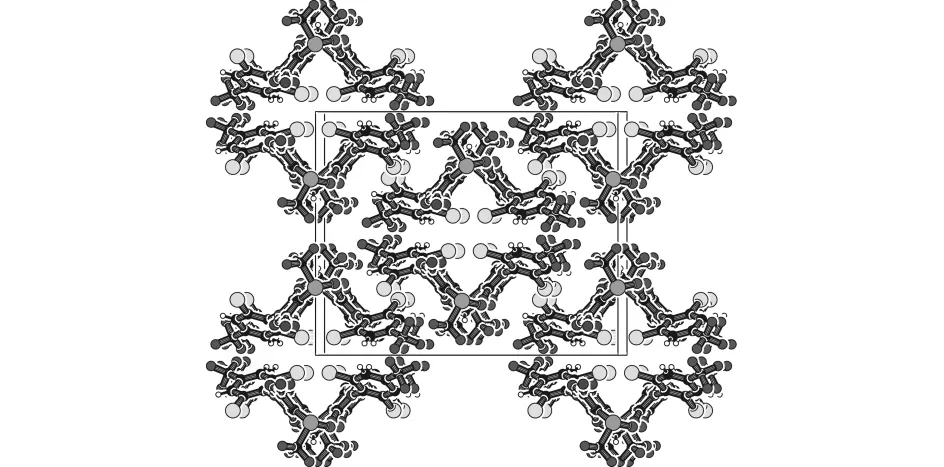
图10 化合物 1c 的结构堆积图Fig.10 Structure stacking diagram of compound 1c

参数数据参数数据分子式C25H8C14F12N8O2S2Z4分子量886.31ρcalc/(g·cm3)1.715T/K293.15μ/mm-15.275晶系单斜F(000)1752空间群P1(21)/c1晶体尺寸/mm30.2×0.2×0.2a/Å20.0584(9)2θ范围/(°)3.517~71.336b/Å16.1373(7)参数范围-22≤h≤24,-11≤k≤19,-12≤l≤13c/Å10.6232(5)收集回响11572α/°90数据/限制/参数3311/0/241β/°93.283(4)F21.024γ/°90最终R指数[I>=2σ(I)]R1=0.0653,wR2=0.1564V/Å33433.1(3)最终R参数(所有数据)R1=0.1156,wR2=0.1971
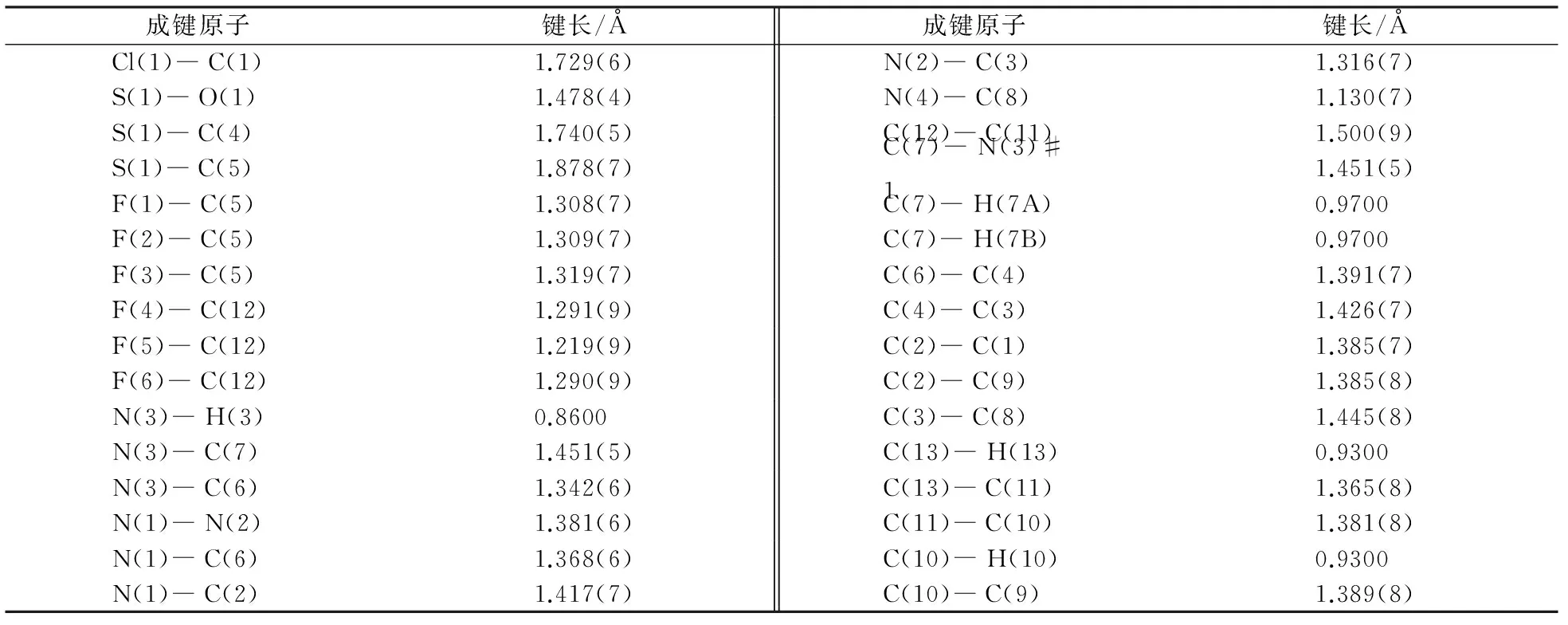
表7 化合物1c的部分键长数据Tab.7 Partial bond length data for compound 1c
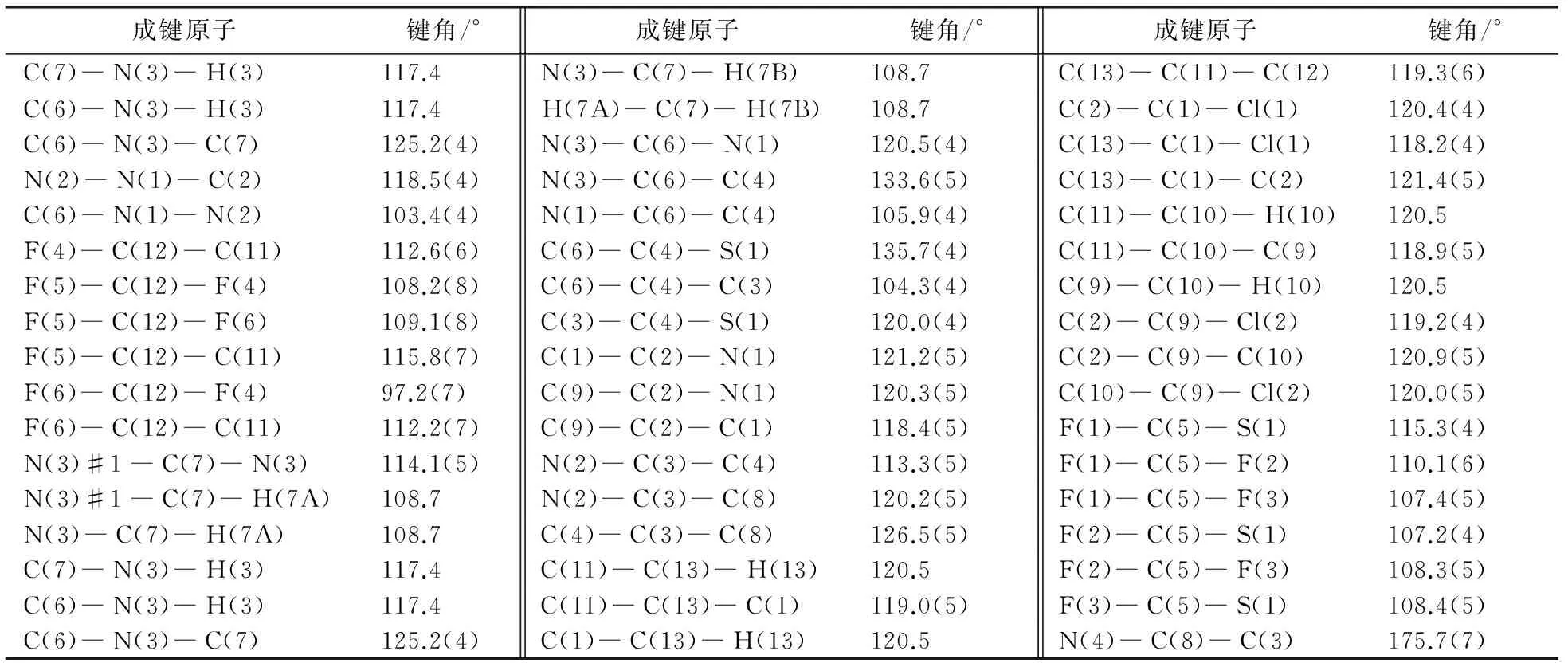
表8 化合物1c的部分键角数据Tab.8 Partial bond angle data for compound 1c
4 结语
本文以芳胺和多聚甲醛为初始原料,使用溶剂热法对芳胺类化合物与甲醛在不同条件和溶剂下进行反应,成功合成了亚甲基二芳胺、三亚甲基三芳胺、四亚甲基四芳胺类衍生物,并利用IR、1H NMR、MS和元素分析表征手段确认了其结构.通过紫外和荧光分析,认为此类亚甲基二芳胺、三亚甲基三芳胺、四亚甲基四芳胺类物质具有较稳定的光物理性能,可用于生物荧光探针、残留物检测等.通过与常规加热回流法比较,发现溶剂热法一锅合成的后续处理简单、操作简便、产率明显提升,是一种一步合成亚甲基二芳胺、三亚甲基三芳胺(氢化均三嗪)和四亚甲基四芳胺的通用方法.
[1] 吕银祥,邹振光,徐 伟,等.亚甲基二胺衍生物的合成新方法及结构表征[J].复旦学报(自然科学版),2007,37(46):286-290.
[2] Desolms S J.N,N,N′,N′-tetramethylmethanediamine.A simple,effective Mannich reagent[J].J Org Chem,1976,41(15): 2650 -2651.
[3] 宋红燕,王 鹏,覃光明,等.一锅法合成二硝基五亚甲基四胺反应机理的研究[J].有机化学,2010,30(3):414-418.
[4] Lu Y X,Liu C M,Zou Z G,et al.Bis(4-phenylpiperazin-1-yl)methane[J].Acta Cryst,2003,59(12):o1960 -o1961.
[5] Dandia A,Arya K,Sati M,Sarawgi P.Green chemical synthesis of fluorinated 1,3,5-triaryl-s-triazines in aqueous medium under microwaves as potential antifungal agents[J].J Fluorine Chem,2004,125(9): 1273-1277.
[6] Giumanini A G,Verardo G,Zangrando E,et al.Revisitation of formaldehyde aniline condensation.VII.1,3,5-Triarylhexahydro-sym-triazines and 1,3,5,7-tetraaryl-1,3,5,7-tetrazocines from aromatic amines and paraformaldehyde[J].Journal für Praktische Chemie,1987,329(6):1087-1103.
[7] Singh A K,Shukla S K,Quraishi M A.Ultrasound mediated green synthesis of hexa-hydro triazines[J].J Mater Environ Sci,2011,2(4) :403-406.
[8] Ghandi M,Salimi F,Olyaei A.Novel reaction ofN,N′-bisarylmethanediamines with formaldehyde.Synthesis of some new 1,3,5-triaryl-1,3,5-hexahydrotriazines[J].Molecules,2006,11:556-563.
[9] 陈连清,周 泉,黄林伟,等.邻菲啰啉缩单胺类席夫碱衍生物及其铕配合物的合成和光物理性能研究[J].中南民族大学学报(自然科学版),2016,35(1):1-6.
[10] 唐和清,阮玉凤,聂 刚,等.溶剂热制备纳米Fe3O4及其高效Fenton降解4-氯苯酚[J].中南民族大学学报(自然科学版),2016,35(2):1-5.
[11] 祁 楠,陈 兴,程侣柏.芳醛与芳胺的缩合反应及产物的倍频效应[J].精细化工,1997,14(1):21-24.
[12] Randaccio L,Zangrando E,Gei M H,Giumanini A G.The cyclic tetramer ofN-methyleneaniline: An X-ray diffraction structure determination[J].Journal für Praktische Chemie,1987,329(2):187-194.
[13] 李卓民.9-芴席夫碱衍生物的合成、晶体结构及性能研究[D].广州: 华南理工大学,2012:37-47.
[14] 金龙飞,范 昕,金呈之,等.L-谷氨酸-5-甲酯金属铜化合物的合成、表征及生物活性[J].中南民族大学学报(自然科学版),2012,31(2):15-19.
One-potSynthesisofMethyleneDiarylamines,TrimethyleneTriarylamineandTetramethyleneTetraarylaminebySolvothermalMethod
ChenLianqing,DuYanting,ZhouQuan,GuoZhiwei
(Key Laboratory of Catalysis and Materials Science of the State Ethnic Affairs Commission & Ministry of Education, College of Chemistry and Materials Science, South-Central University for Nationalities, Wuhan 430074,China)
In order to find a more efficient and simple method for the synthesis of methylene aromatic amines, a series of methylene diarylamine, trimethylene triarylamine; and tetramethylene tetraarylamine compounds were one-pot synthesized by solvothermal method using arylamine and paraformaldehyde as starting materials in different solvents under different conditions.Their structures were characterized by IR,1H NMR, elemental analysis and so on.The effects of different substituents on benzene ring and reaction time on the product were also investigated.The results indicated that the solvothermal method had the advantages of simple post-treatment and high yield.It is a new general method for the synthesis of methylene diarylamine, triamine and tetramine derivatives with good photoluminescence properties.
aromatic amines; paraformaldehyde; solvothermal method; methylene diarylamine; trimethylene triarylamine; tetramethylene tetraarylamine
2017-04-20
陈连清(1979-),男,副教授,博士,研究方向:有机合成,E-mail: lqchen@mail.scuec.edu.cn
国家自然科学基金资助项目(20702064, 21177161, 31402137);人事部留学人员科技活动择优资助项目(BZY13007);湖北省杰出青年基金资助项目(2013CFA034);湖北省青年英才开发计划(RCJH15001);绿色催化四川省高校重点实验室开放课程基金项目(LYJ1107)
O626.4
A
1672-4321(2017)04-0001-08
猜你喜欢
矿产保护与利用(2022年2期)2023-01-09
声屏世界(2022年15期)2022-11-08
分子催化(2022年1期)2022-11-02
化学工业与工程(2022年1期)2022-03-29
食品安全导刊(2021年21期)2021-08-30
中成药(2019年12期)2020-01-04
校园英语·中旬(2019年5期)2019-07-16
大自然探索(2016年3期)2016-04-07
中国报道(2015年3期)2015-09-10
火炸药学报(2014年3期)2014-03-20
