
蛋白激酶C参与大鼠慢性偏头痛中枢敏化*
2017-11-20 03:01吴白雪秦光成谢景梅周冀英陈力学
中国疼痛医学杂志 2017年5期
吴白雪 王 莎 秦光成 肖 垚 谢景梅 谭 戈 周冀英 陈力学Δ
(1重庆医科大学附属第一医院实验研究中心,重庆 400016;2重庆市神经病学重点实验室,重庆 400016)
·论 著·
蛋白激酶C参与大鼠慢性偏头痛中枢敏化*
吴白雪1王 莎1秦光成1肖 垚1谢景梅1谭 戈2周冀英2陈力学1Δ
(1重庆医科大学附属第一医院实验研究中心,重庆 400016;2重庆市神经病学重点实验室,重庆 400016)
目的:本文旨在研究蛋白激酶C (PKC)是否通过中枢敏化参与慢性偏头痛病理生理过程。方法:SD雄性大鼠随机分为7组,假手术组(n= 11),模型组 (n= 12),模型+溶剂组(n= 8),PKC抑制剂4 µg组(n= 11),PKC抑制剂8 µg组(n= 10),PKC激动剂100 ng组(n= 10),PKC激动剂200 ng组(n= 11)。“炎性汤”反复刺激硬脑膜,模拟硬脑膜痛觉感受器的反复激活,建立慢性偏头痛大鼠模型,侧脑室给予PKC抑制剂和PKC激动剂,使用von Frey test测定大鼠面部及后足的机械痛阈值,Western blot 检测PKC、CGRP、c-Fos蛋白的表达变化。结果:“炎性汤”反复刺激硬脑膜后,大鼠面部及足底的机械痛阈值显著降低,而三叉神经脊束尾核部位的CGRP、c-Fos及PKC蛋白表达显著增加。使用CHE抑制PKC可以显著增高大鼠面部及足底痛阈值,显著降低CGRP、c-Fos的表达;而使用PMA激活PKC则降低痛阈值,显著增高CGRP、c-Fos的表达。结论:本研究表明PKC可以调节机械痛阈值以及CGRP、c-Fos的表达变化,提示PKC参与慢性偏头痛中枢敏化的病理生理过程。
蛋白激酶C;慢性偏头痛;中枢敏化;三叉神经脊束尾核
偏头痛是一种临床常见的反复发作原发性头痛疾病,根据其发作频率可分为发作性偏头痛(episodic migraine, EM)和慢性偏头痛(chronic migraine, CM)。慢性偏头痛在普通人群中的患病率约为 2%,给患者和社会均带来了严重的经济负担,已经被 WHO列为四种最严重的慢性功能障碍性疾病之一[1]。然而目前尚无针对慢性偏头痛的特效治疗方法和根治手段,因此慢性偏头痛的病理机制研究对于探寻新的治疗方法至关重要。目前有许多研究表明中枢敏化(central sensitization)这一机制在慢性偏头痛的发病机制中起着尤为重要的作用[2~5]。偏头痛中枢敏化主要是由三叉神经脊束尾核(trigeminal nucleus caudalis, TNC)部位神经元的兴奋性异常增高引起,从而导致皮肤痛觉过敏(hyperalgesia )和痛觉超敏(cutaneous allodynia, CA)[6,7]。蛋白激酶C (protein kinase C, PKC)是重要的细胞内信号转导分子,广泛分布于中枢神经系统[8,9],已有研究证实在疼痛的中枢调节中,PKC可能起着关键性的作用,抑制PKC可以改善中枢敏化,减轻痛觉过敏[9,10]。然而关于PKC是否参与慢性偏头痛的中枢敏化机制,目前尚无相关报道。因此,本实验参照Melo-Carrillo报道的方法建立大鼠慢性偏头痛模型[11],检测TNC部位PKC的蛋白表达量,然后给予PKC的激动剂Phorbol 12-myristate 13-acetate (PMA)和抑制剂chelerythrine chloride (CHE),应用Von Frey test检测面部和足底的机械痛阈值,Western blotting检测TNC部位CGRP、c-Fos的蛋白表达水平,探讨PKC在慢性偏头痛的中枢敏化机制中的作用,以期待为慢性偏头痛的防治提供新的靶点。
方 法
1.实验动物
清洁级 SD 雄性大鼠(SPF)73只(体重250~300 g),由重庆医科大学实验动物中心提供(实验动物生产许可证号:SYXK(渝)2012-0001),实验动物均置于22±2 ℃室温,湿度50±5%以及12 h/12 h 昼夜明暗交替控光的环境中,自由摄食饮水。所有实验SD大鼠适应性喂养一周后进入实验。本研究均遵守国际疼痛学会(IASP)相关指南,尽量减少实验动物数目,尽量降低对实验动物的伤害,并经重庆医科大学附属第一医院伦理委员会的许可。
2.慢性偏头痛中枢敏化模型的建立
参照 Melo-Carrillo等人的报道:通过“炎性汤”(inflammatory soup, IS)反复刺激硬脑膜建立慢性偏头痛模型[11]。腹腔注射 10%的水合氯醛(i.p.0.4 g/kg体重)麻醉后,将大鼠固定于立体定位仪(美国 Stoelting公司)上,常规备皮、消毒。头部正中切开约 1 cm的皮肤切口,钝性分离皮下组织,3% H2O2处理颅骨表面,台式颅骨钻建立一个孔径约1 mm 的颅骨窗,避免损伤硬脑膜。在骨窗安装微量套管,使用自凝牙托粉和自凝牙托水固定,全层缝合皮肤,术后将大鼠置于37 ℃恒温板,待苏醒后送回饲养间,每只实验大鼠在同等条件下分笼饲养。术后1周后,选择伤口未感染,恢复良好的SD大鼠进入实验,随机分到不同的实验组。参照文献报道的“炎症汤”配方:组胺(1 mmol/L)、5-羟色胺(1 mmol/L)、缓激肽(1 mmol/L)和前列腺素E2(0.1 mmol/L)加入PBS缓冲液配制而成[11,12]。每日通过微螺旋定量推进装置匀速缓慢泵入2 µl“炎症汤”滴注到硬脑膜表面,重复7天;Sham 组大鼠在同样的时间给予PBS硬脑膜滴注。
3.侧脑室注射
本实验使用的PKC抑制剂CHE (chelerythrine chloride, Abcam)及PKC激 动 剂 PMA (Phorbol 12-myristate 13-acetate, Abcam)在既往文献[12~17]中证实对PKC有明确的抑制或激动作用,此次试验中CHE(10 mmol/L)及PMA(100 mmol/L)溶于DMSO,经侧脑室给药,PKC抑制剂低剂量组给予CHE(4 µg/只),PKC抑制剂高剂量组给予CHE(8µg/只),PKC激动剂低剂量组给予PMA(100 ng/只),PKC激动剂高剂量组给予PMA(200 ng/只)。建模成功后(7次“炎性汤”滴注硬脑膜),麻醉好的SD 大鼠固定在立体定位仪上,常规消毒、备皮,切开皮下组织,止血钳取出微量套管,3%H2O2处理颅骨表面,在前囟左侧1.5 mm后方1 mm,颅骨钻建立直径约1 mm骨窗。微量注射器固定于立体定位仪上,将微量注射器的针头从颅骨窗插入约 4 mm 到达侧脑室后,匀速缓慢泵入药物5 µl(10 min)。待药物注射完成,留针 5 min 后拔出,用骨蜡封闭骨窗,缝合皮肤。SD大鼠置于 37 ℃恒温板,苏醒后送回饲养间。
4.机械痛阈值的测定
机械性痛阈值的测定参照Oshinsky报道的方法[18]。将实验SD大鼠置于22 cm×22 cm×30 cm的透明笼子中适应10 min后,使用电子Von Frey痛阈仪(WoodLand Hills,CA,USA)测定大鼠面部、足底机械痛阈值。用痛阈仪的硬头垂直轻触大鼠面部或后爪的皮肤,大鼠头部或后爪快速回缩,仪器自动记录痛阈值,每个部位测 3 次,然后取平均值作为该部位的疼阈值。每次给予炎性汤前测定其基础痛阈值,以及给予PKC激动剂和抑制剂24小时后,再次测定各组痛阈值的改变。
5.蛋白提取与蛋白水平检测
将大鼠麻醉后断头取脑,取三叉神经脊束尾核(TNC)组织,加入含有PMSF (Beyotime,China)和磷酸酶抑制剂(Boster, China)的RIPA缓冲液(Beyotime, China),充分匀浆后置于4 ℃冰箱90 min,使其充分裂解,然后,将组织液放入低温离心机离心(4 ℃,12 000 rpm, 20 min),取上清液,用BCA试剂盒(Beyotime, China)测定蛋白浓度。待测样品加入SDS-PAGE蛋白上样缓冲液(Beyotime,China),放入100 ℃沸水使其变性 5 min,冷却后分装。取50 µg/孔蛋白样品上样,10% SDS-PAGE胶(Beyotime,China)电泳分离后,电转(250 mA)至PDVF膜,5%脱脂奶粉室温封闭2小时。分别加入相应一抗anti-PKC (1:1 000, Abcam)、anti-CGRP(1:2 000, abcam)和anti-c-Fos (1:500, Abcam) 4℃孵育过夜。第二日复温2小时,TBST 漂洗 3 次,10 min/次。加入相应浓度二抗(1:5 000,北京中杉金桥生物技术有限公司,China)37 ℃孵育2小时。TBST 漂洗 3 次,10 min/次。采用ECL (Beyotime,China)试剂盒和凝胶成像系统(Fusion, Germany)发光成像。灰度值结果使用Fusion软件分析。
6.数据处理与统计分析
计量资料均以均值±标准误(±SEM)表示,使用统计软件SPSS 19.0进行统计学分析。两组之间的均值比较采用独立样本t检验(independentsamples T test),多组均值的比较采用单因素方差分析(ANOVA),P< 0.05认为差异有统计学意义。
结 果
1.“炎性汤”反复刺激硬脑膜后,大鼠面部及足底的机械痛阈值显著降低,而三叉神经脊束尾核部位CGRP、c-Fos及PKC的蛋白表达显著增加
用电子Von Frey痛阈仪测定大鼠面部、足底机械痛阈值结果发现:与给药前的面部及足底基础痛阈值 (7.25±0.35 g;15.90±0.44 g)比较,反复硬膜滴注PBS组大鼠机械痛阈值(7.75±0.30 g;15.00±0.34 g)无 明 显 统 计 学 差 异 (P> 0.05);而反复硬膜滴注IS组大鼠的面部及足底痛阈值(3.09±0.20 g;5.89±0.42 g)与 PBS组 的 痛 阈 值(7.75±0.30 g;15.00±0.34 g)相比,显著下降(P<0.05, 见图 1、2)。Western blot结果显示:硬膜外滴注7次“炎性汤”后,CM组大鼠TNC部位CGRP、c-Fos的蛋白表达较Sham组均显著增高(P<0.05)。以上结果表明反复硬膜外滴注炎性汤可以降低大鼠机械痛阈值,导致TNC部位CGRP、c-Fos表达增高,提示该模型成功模拟了慢性偏头痛的中枢敏化机制。此外,CM组TNC部位PKC蛋白表达水平较Sham组亦显著增高(P< 0.05),该结果表明了PKC可能在慢性偏头痛中起着重要作用。
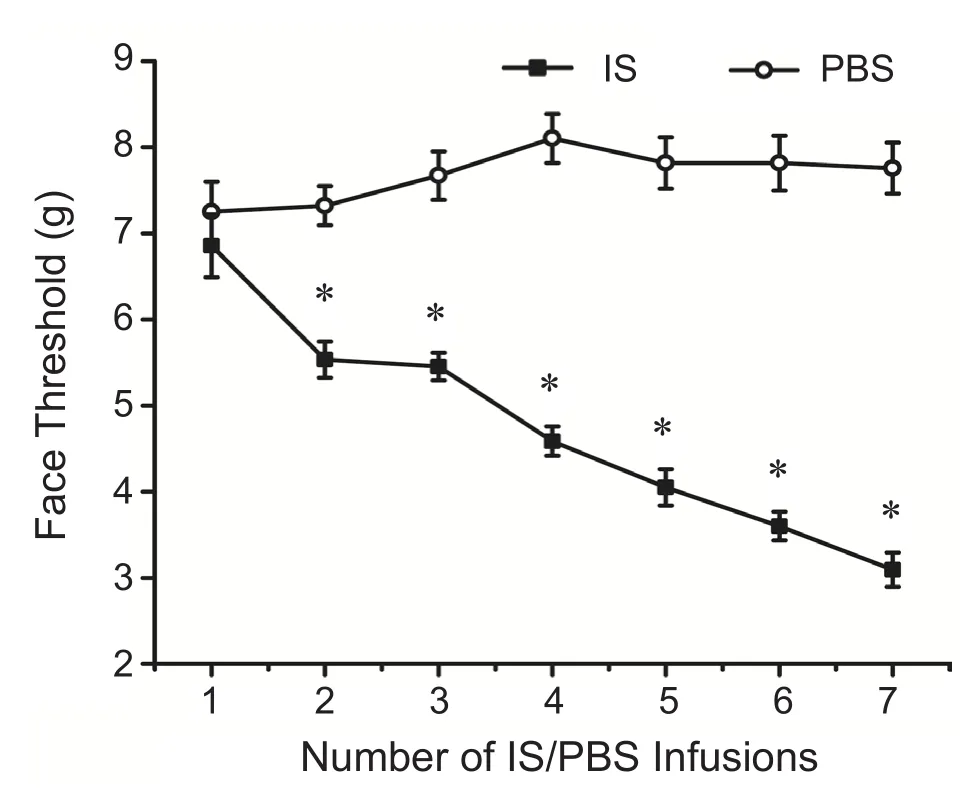
图1 两组大鼠滴注不同IS/PBS次数的面部机械痛阈值的改变情况(n=16,±SEM)*P < 0.05,与 PBS 组相比Fig.1 The change of mechanical threshold in face after IS/PBS infusion in each group (n=16,±SEM)* P < 0.05, compared with PBS group

图2 两组大鼠滴注不同IS/PBS次数的足底机械痛阈值的改变情况(n=16,±SEM)*P < 0.05,与 PBS 组相比Fig.2 The change of mechanical threshold in hindpaw after IS/PBS infusion in each group (n=16,±SEM)*P < 0.05, compared with PBS group.
2.使用CHE抑制PKC可以显著增高大鼠面部及足底痛阈值,而使用PMA激活PKC则降低大鼠面部及足底痛阈值

图3 给予PKC抑制剂CHE后各组大鼠面部机械痛阈值的比较*P < 0.05,与 Sham 组相比;#P < 0.05,与 CM+DMSO 组相比Fig.3 The comparison of mechanical threshold in face among different groups after administration of PKC inhibitor CHE*P < 0.05, compared with Sham group; #P < 0.05,compared with CM+DMSO group.

图5 给予PKC抑制剂CHE后各组大鼠足底机械痛阈值的比较*P < 0.05,与 Sham组相比; #P < 0.05,与 CM+DMSO 组相比Fig.5 The comparison of mechanical threshold in hindpaw among different groups after administration of PKC inhibitor CHE*P < 0.05, compared with Sham group; #P < 0.05, compared with CM+DMSO group.
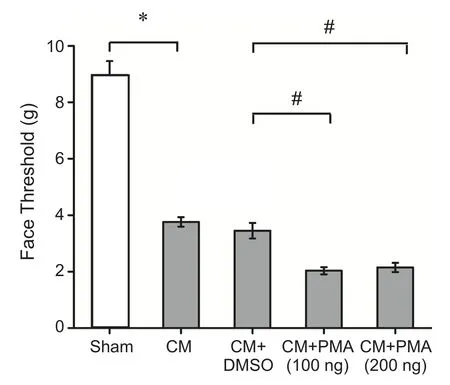
图4 给予PKC激动剂PMA后各组大鼠面部机械痛阈值的比较*P < 0.05,与Sham 组相比; #P < 0.05,与 CM+DMSO组相比Fig.4 The comparison of mechanical threshold in face among different groups after administration of PKC activator PMA*P < 0.05, compared with Sham group; #P < 0.05, compared with CM+DMSO group.
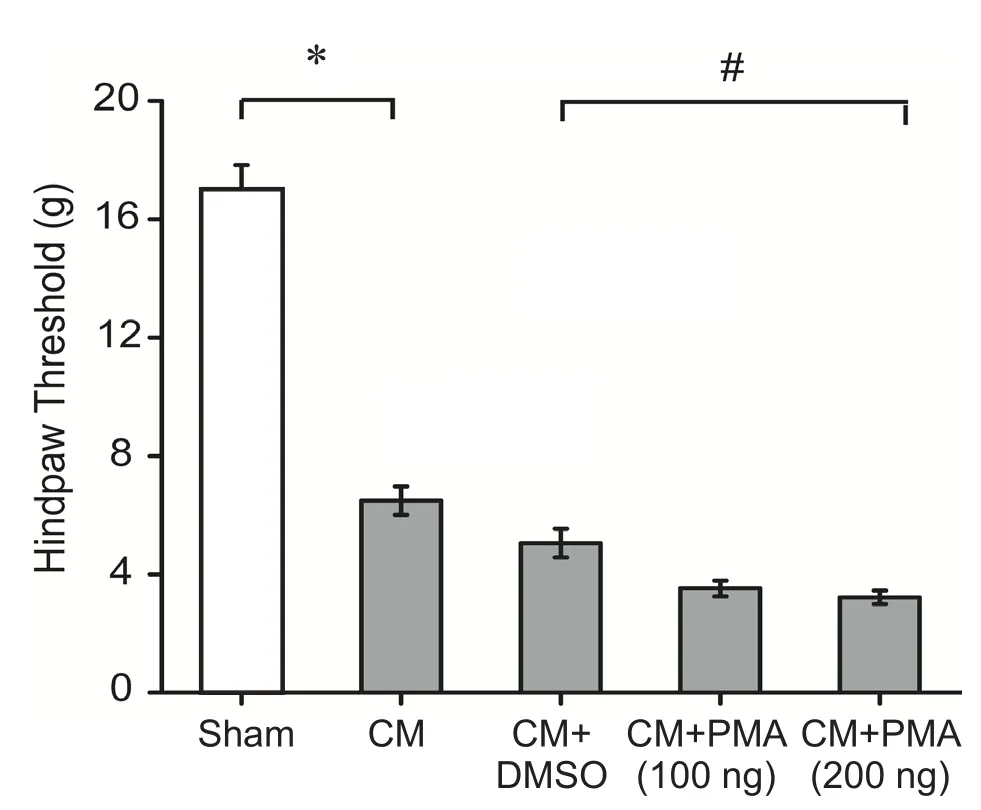
图6 给予PKC激动剂PMA后各组大鼠足底机械痛阈值的比较*P < 0.05,与 Sham 组相比;#P < 0.05,与 CM+DMSO 组相比Fig.6 The comparison of mechanical threshold in hindpaw among different groups after administration of PKC activator PMA*P < 0.05, compared with Sham group; #P < 0.05, compared with CM+DMSO group.
与Sham组面部及足底机械痛阈值(8.97±0.50 g;17.02±0.82 g)比较,CM组(3.76±0.17 g;6.49±0.48 g)及 CM+DMSO 组 (3.45±0.28 g;5.05±0.48 g)机械痛阈值均显著降低(P< 0.05),而CM组与CM+DMSO组大鼠面部及足底痛阈值并无统计学差异(P> 0.05),提示侧脑室给予DMSO并不影响大鼠的机械痛阈值。给予PKC抑制剂CHE后,大鼠面部及足底机械痛阈值均显著增高(P< 0.05),但是,CHE高剂量组(6.38±0.38 g;10.36±0.75 g)与低剂量组(5.19±0.31 g;8.80±0.60 g)面部及足底机械痛阈值并无明显统计学差异(P> 0.05, 见图3、4),提示CHE改善痛觉过敏不成剂量依耐性。此外,给予PKC激动剂PMA后,高、低剂量组大鼠面部机械痛阈值(2.14±0.17 g;2.03±0.13 g)均显著降低(P< 0.05),但是,PMA低剂量组(3.52±0.27 g)的大鼠足底痛阈值变化较CM组无统计学差异,而PMA高剂量组的足底痛阈值(3.22±0.23 g)则显著下降(P< 0.05, 见图5、6),PMA高、低剂量组之间面部及足底机械痛阈值亦无明显统计学差异,提示PMA加重面部痛觉过敏不成剂量依赖性,以及高剂量的PMA可以降低大鼠足底的痛阈值。
3.使用CHE抑制PKC可以显著降低CGRP、c-Fos的表达,而使用PMA激活PKC则显著增高CGRP、c-Fos的表达
与Sham组相比,CM组TNC部位CGRP的表达显著增高(P< 0.05);给予PKC抑制剂CHE后,CGRP的表达显著降低(P< 0.05);而给予PKC激动剂PMA后,CGRP的表达显著增高(P< 0.05)。此外,与Sham组比较,CM组TNC部位c-Fos的表达量亦显著增高(P< 0.05);给予PKC的抑制剂CHE后,c-Fos的表达显著降低(P< 0.05);而给予PKC的激动剂后,c-Fos的表达显著增高 (P< 0.05, 见图 7~9)。

图7 两组PKC蛋白表达量*P < 0.05,与 Sham 组相比Fig.7 The comparison of PKC protein expression levels between two groups*P < 0.05, compared with sham group.
讨 论
每年大约有2%的发作性偏头痛转化为慢性偏头痛[19]。慢性偏头痛因为频繁和严重的头痛发作影响患者的学习与工作能力、生活质量, 给患者个人以及社会带来了非常严重的危害。因此,治疗及预防慢性偏头痛是神经病学的一重要领域。在偏头痛的病理过程,尤其是偏头痛慢性化过程中,中枢敏化发挥了重要作用[5~7]。Burstein R等人1996年首次在Nature杂志上提出中枢敏化参与了偏头痛发病过程[20]。中枢敏化主要是由于中枢神经系统在疼痛产生以及疼痛传递的过程中发生了可塑性变化,从而导致中枢神经系统对伤害区域内和伤害区域周围非伤害区域的刺激反应增强(痛觉过敏,hyperalgesia),对非伤害性刺激产生疼痛异常的伤害性反应(痛觉超敏,allodynia)[21]。皮肤异常性疼痛(CA)作为偏头痛中枢敏化的外在征象,疼痛范围牵涉面部,还可以扩展到整个面部以及头皮,甚至达到四肢[22~25]。在偏头痛的动物模型中,多采用电、化学刺激敏化硬脑膜以及三叉神经血管系统来诱导这种中枢敏化状态,多以 von-Frey 丝检测面部和后足的皮肤痛阈来评价[26]。本研究发现,与对照组(PBS组)相比,慢性偏头痛组大鼠(IS组)面部及足底的机械痛阈值都显著降低,随着“炎性汤”刺激硬脑膜次数增加,大鼠面部及足底痛阈值进行性下降,提示“炎性汤”反复刺激硬脑膜可以诱导皮肤异常性疼痛(CA),导致偏头痛的中枢敏化状态。
降钙素基因相关肽(calcitonin gene related peptide,CGRP)是一种血管舒张肽,广泛分布于中枢系统,是三叉神经微血管激活的标志物,在偏头痛中枢敏化的疼痛感觉调控中发挥着重要的作用[27,28]。Cady等的研究表明CGRP可以促进中枢以及外周的三叉神经元以及神经胶质细胞的敏感性[29]。c-Fos是即刻早期基因的重要成员,参与偏头痛的痛觉调控过程[7,30]。CGRP 和c-Fos在偏头痛病理过程,尤其在慢性偏头痛的发展和维持中起重要作用,可以作为中枢敏化和神经元激活的生物学标志[7,27~30]。本研究发现:慢性偏头痛大鼠(7次“炎性汤”后)TNC部位观察到CGRP和c-Fos表达的显著增加,提示重复给予“炎性汤”刺激硬脑膜可以导致中枢敏化和神经元激活。综上所述,在我们的研究中:给予“炎性汤”反复刺激硬脑膜可以显著降低大鼠面部和足底痛阈值,增加中枢敏化标志CGRP 和c-Fos的表达,提示“炎性汤”反复刺激硬脑膜可以较好的模拟慢性偏头痛中枢敏化的过程,该模型是一种可行的偏头痛中枢敏化模型。
蛋白激酶C(PKC)是重要的细胞内信号转导分子,至今已发现12种亚型,它们构成了一个丝氨酸/苏氨酸激酶超家族。PKC在介导细胞分裂、增殖、凋亡、细胞骨架蛋白的重塑、离子通道的调节及细胞分泌等方面发挥着重要作用[8,9]。既往的研究发现,在多种疼痛模型中, PKC活性显著增高,而抑制PKC的活性后往往可以明显减轻疼痛,提示PKC在疼痛的中枢敏化形成过程中发挥着重要作用[3,9,10]。本研究结果发现,大鼠慢性偏头痛中枢敏化模型中,观察到伴随着大鼠面部及足底痛阈值的下降,TNC部位PKC的蛋白表达水平显著增加,而使用PKC的抑制剂CHE后可以显著改善CM大鼠痛觉过敏,进一步使用PKC激动剂PMA可以加重大鼠痛觉过敏,提示PKC同样参与慢性偏头痛痛觉过敏的过程。前文提到:CGRP 和c-Fos可以作为偏头痛中枢敏化和神经元激活的生物学标志。在此次研究中发现,大鼠慢性偏头痛中枢敏化模型中,使用PKC的抑制剂CHE后可以显著抑制CGRP 和c-Fos的表达,而使用PKC激动剂PMA可以增加CGRP 和c-Fos的表达,提示PKC参与了慢性偏头痛中枢敏化的病理生理过程。PKC共有12 种亚型,已有研究表明PKC γ亚基是痛觉过敏、中枢敏化关键的调节因子[31~33],遗憾的是,此次研究并未细究PKC各亚型对偏头痛中枢敏化的作用,这将是我们下一步研究的重点。
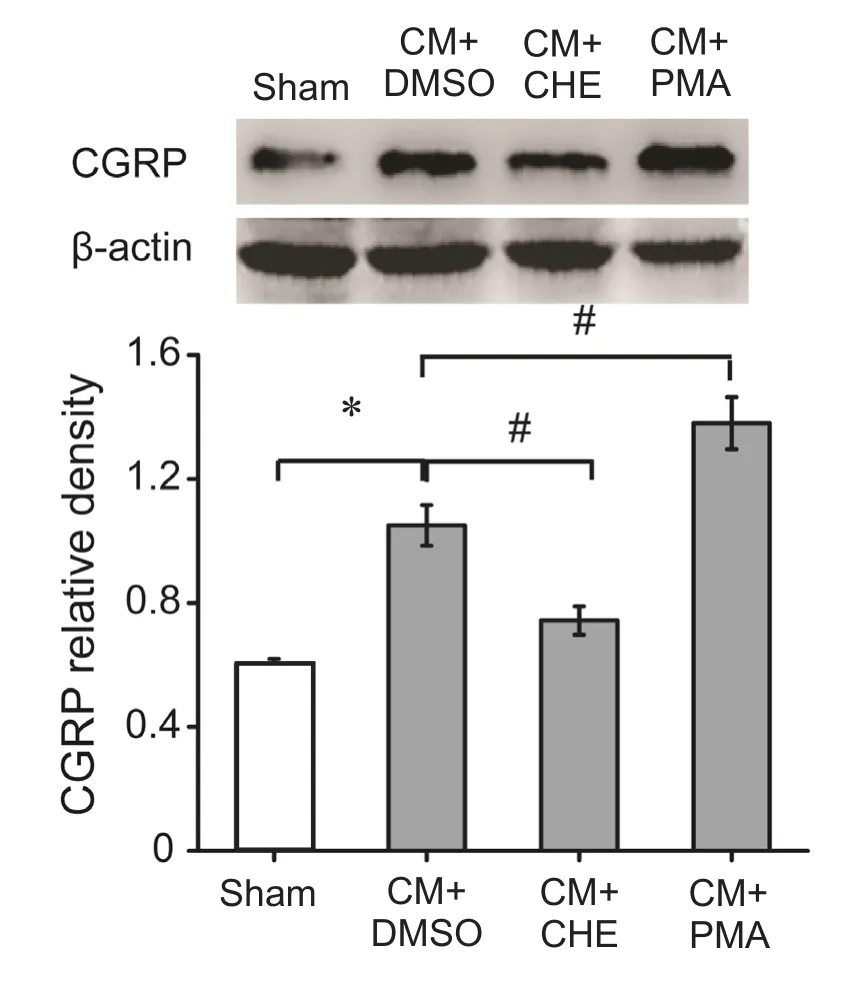
图8 各组CGRP蛋白表达量*P < 0.05,与Sham组相比; #P < 0.05,与CM+DMSO组相比Fig.8 The comparison of CGRP protein expression levels among four groups*P < 0.05, compared with Sham group; #P < 0.05, compared with CM+DMSO group.
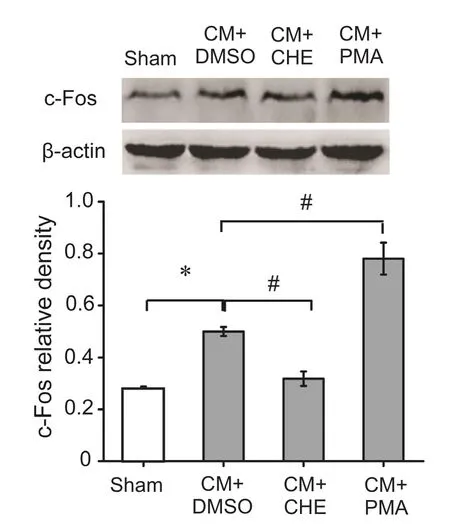
图9 各组c-Fos蛋白表达量*P < 0.05,与Sham组相比;#P < 0.05,与CM+DMSO组相比Fig.9 The comparison of c-Fos protein expression levels among four groups*P < 0.05, compared with Sham group;#P < 0.05, compared with CM+DMSO group.
综上所述,“炎性汤”反复刺激硬脑膜后,大鼠面部及足底机械痛阈值显著降低,CGRP、c-Fos表达显著增加,证实了该模型是一种可行的慢性偏头痛中枢敏化模型。此外,PKC可以有效调节中枢敏化标志CGRP、c-Fos的表达变化及大鼠机械痛阈值的变化,提示PKC参与慢性偏头痛中枢敏化的病理生理过程。我们的研究不仅为慢性偏头痛的动物模型进行了探索证实,更为未来慢性偏头痛的预防治疗提供了可靠思路。
[1]Schwedt TJ. Chronic migraine. BMJ, 2014, 348(12):g1416.
[2]郑颖, 张伟, 朱昱,等. 慢性偏头痛的研究进展. 国际神经病学神经外科学杂志, 2012, 39(4):327 ~ 330.
[3]Burstein R, Jakubowski M, Rauch SD. The science of migraine. J Vestib Res, 2011, 21(6):305 ~ 314.
[4]Burstein R. Deconstructing migraine headache into peripheral and central sensitization. Pain, 2001, 89(2-3):107 ~ 110.
[5]David Dodick MD, Stephen Silberstein MD. Central Sensitization Theory of Migraine: Clinical Implications.Headache, 2006, 46 Suppl 4(Supplement s4): S182 ~S191.
[6]Aguggia M. Allodynia and migraine. Neurol Sci, 2012,33(1):9 ~ 11.
[7]Boyer N, Dallel R, Artola A,et al. General trigeminospinal central sensitization and impaired descending pain inhibitory controls contribute to migraine progression. Pain, 2014, 155(7):1196 ~ 1205.
[8]倪同尚, 武胜昔, 李云庆. 蛋白激酶C的分子生物学特征及研究进展. 解剖科学进展, 2001, 7(4):335 ~ 338.
[9]Velázquez KT, Mohammad H, Sweitzer SM. Protein kinase C in pain: Involvement of multiple isoforms.Pharmacol Res, 2007, 55(6):578 ~ 589.
[10]王远胜, 胡兴国. 蛋白激酶C与疼痛的中枢敏感化.国际麻醉学与复苏杂志, 2002, 23(2):108 ~ 111.
[11]Melo-Carrillo A, Lopez-Avila A. A chronic animal model of migraine, induced by repeated meningeal nociception,characterized by a behavioral and pharma-cological approach. Cephalalgia, 2013, 33(13): 1096 ~ 1105.
[12]Begon S, Pickering G, Eschalier A,et al. Role of spinal NMDA receptors, protein kinase C and nitric oxide synthase in the hyperalgesia induced by magnesium de fi ciency in rats. Bri J Pharmacol, 2001, 134(6):1227 ~1236.
[13]Li SJ, Cui SY, Zhang XQ,et al. PKC in rat dorsal raphe nucleus plays a key role in sleep-wake regulation. Prog Neuro Psychopharmacol Biol Psychiatry, 2015, 63:47 ~ 53.
[14]Hsieh WK, Lin HH, Lai CC. Involvement of protein kinase C and Src tyrosine kinase in acute tolerance to ethanol inhibition of spinal NMDA-induced pressor responses in rats. Br J Pharmacol, 2009, 158(3):806 ~818.
[15]Shibasaki M, Mizuno K, Kurokawa K,et al.L-type voltage-dependent calcium channels facilitate acetylation of histone H3 through PKCγ phosphorylation in mice with methamphetamine-induced place preference. J Neurochem, 2011, 118(6):1056.
[16]Zhang Y, Gong K, Zhou W,et al. Involvement of Subtypes γ and of Protein Kinase C in Colon Pain Induced by Formalin Injection. Neurosignals, 2011,19(3):142.
[17]Coderre TJ. Contribution of protein kinase C to central sensitization and persistent pain following tissue injury.Neurosci Lett, 1992, 140(2):181 ~ 184.
[18]Oshinsky ML, Gomonchareonsiri S. Episodic Dural Stimulation in Awake Rats: A Model for Recurrent Headache. Headache, 2007, 47(7):1026 ~ 1036.
[19]Bigal ME, Serrano D, Buse D,et al. Acute migraine medications and evolution from episodic to chronic migraine: a longitudinal population-based study. Headache,2008, 48:1157 ~ 1168.
[20]Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature, 1996, 384(6609):560 ~ 564.
[21]Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science, 2000, 288(5472):1765 ~ 1769.
[22]Jakubowski M, Silberstein S, Ashkenazi A,et al. Can allodynic migraine patients be identi fi ed interictally using a questionnaire?. Neurology, 2005, 65(9):1419 ~ 1422.
[23]Ashkenazi A, Silberstein S, Jakubowski M,et al.Improved identi fi cation of allodynic migraine patients using a questionnaire. Cephalalgia, 2007, 27(4):325 ~329.
[24]Mathew NT, Kailasam J, Seifert T. Clinical recognition of allodynia in migraine. Neurology, 2004, 63(5):848 ~852.
[25]裴培, 刘璐, 赵洛鹏,等. 偏头痛:中枢敏化与皮肤异常性疼痛. 中国疼痛医学杂志, 2016, 22(2):128 ~132.
[26]Storer RJ, Supronsinchai W, Srikiatkhachorn A. Animal models of chronic migraine. Curr Pain Headache Rep,2015, 19:467.
[27]谭亮, 樊光辉. 偏头痛发病机制的研究进展. 中国临床神经外科杂志, 2012, 29(9):14 ~ 15.
[28]Durham PL. Diverse Physiological Roles of Calcitonin Gene-Related Peptide in Migraine Pathology: Modulation of Neuronal-Glial-Immune Cells to Promote Peripheral and Central Sensitization. Curr Pain Headache Rep, 2016,20(8):1 ~ 9.
[29]Cady RJ, Glenn JR, Smith KM,et al. Calcitonin gene related peptide promotes cellular changes in trigeminal neurons and glia implicated in peripheral and central sensitization. Mol Pain, 2011, 7(1): 94.
[30]Mitsikostas DD, Rio MSD. Receptor systems med-iating c-fos, expression within trigeminal nucleus caudalis in animal models of migraine. Brain Res Brain Res Rev,2001, 35(1):20 ~ 35.
[31]Xie HY, Fei X, Yue L,et al. Increases in PKC gamma expression in trigeminal spinal nucleus is associated with orofacial thermal hyperalgesia in streptozotocin-induced diabetic mice. J Chem Neuroanat, 2015, 63:13 ~ 19.
[32]Hughes AS, Averill S, King VR,et al. Neurochemical characterization of neuronal populations expressing protein kinase C gamma isoform in the spinal cord and gracile nucleus of the rat. Neuroscience, 2008, 153(2):507 ~ 517.
[33]Malmberg AB, Chen C, Tonegawa S,et al. Preserved acute pain and reduced neuropathic pain in mice lacking PKCgamma. Science, 1997, 278(5336):279 ~ 283.
PKC CONTRIBUTES TO CENTRAL SENSITIZATION IN A RAT MODEL OF CHRONIC MIGRIANE*
WU Bai-Xue1, WANG Sha1, QIN Guang-Cheng1, XIAO Yao1, XIE Jing-Mei1, TAN Ge2, ZHOU Ji-Ying2,CHEN Li-Xue1Δ
(1Laboratory Research Center, The First Af fi liated Hospital of Chongqing Medical University, Chongqing 400016, China;2Chongqing Key Laboratory of Neurology, Chongqing 400016, China)
Objective: The purpose of this paper is to investigate whether PKC participates in central sensitization in chronic migraine pathophysiological process. Methods: Sprague-Dawley rats were randomly divided into 7 groups: Sham group (n=11), CM group (n=12), CM+DMSO group (n=8), CM+CHE4 µg group (n=11), CM+CHE 8 µg group (n=10), CM+PMA100 ng group (n=10), CM+PMA 200 ng group(n=11). We repeatedly infused inflammatory soup (IS) on the intact dura in awake rats to model the recurrent dural nociceptor activation assumed to occur in patients with chronic migraine (CM). PKC blocker chelerythrine chloride (CHE) and PKC activator Phorbol 12-myristate 13-acetate (PMA) were administrated to investigate the role of PKC in central sensitization induced by in fl ammatory soup. Then we used von Frey test to detect the change of pain threshold. Western blotting was performed to detect the expression of PKC,CGRP and c-Fos in trigeminal nucleus caudalis (TNC). Results: Repeated infusion of IS decreased pain threshold in the face and hindpaw region, and up-regulated the expression of PKC, CGRP and c-Fos in TNC.Furthermore, inhibition of PKC by CHE relieved the allodynia, and reduced the expression of CGRP and c-Fos. Activation of PKC by PMA aggravated the allodynia, and increased the expression of CGRP and c-Fos.Conclusion: Inhibition and activation of PKC could regulate the pain threshold and the expression of CGRP and c-Fos. These data indicate that PKC might play a prominent part in central sensitization of CM model.
PKC; Chronic migraine; Central sensitization; Trigeminal nucleus caudalis
10.3969/j.issn.1006-9852.2017.05.004
国家自然科学基金(No. 81671093, No. 81500957);重庆市渝中区科技计划项目(YCSTC,2016BE02)
△通讯作者 chenlixue@hospital.cqmu.edu.cn
猜你喜欢
轻金属(2022年2期)2022-06-16
世界科学技术-中医药现代化(2022年2期)2022-05-25
昆明医科大学学报(2021年12期)2021-12-30
昆明医科大学学报(2021年4期)2021-07-23
化工设计通讯(2021年2期)2021-01-07
陶瓷学报(2020年3期)2020-10-27
辽宁省博物馆馆刊(2020年0期)2020-08-13
世界最新医学信息文摘(2020年52期)2020-07-09
人大建设(2019年12期)2019-05-21
中医眼耳鼻喉杂志(2019年2期)2019-04-13
