
高通量测序分析肠易激综合征患者和健康人肠道菌群差异*
2017-11-07 07:29汕头大学医学院广东汕头515041
罕少疾病杂志 2017年5期
1.汕头大学医学院 (广东 汕头 515041)
2.北京大学深圳医院消化内科 (广东 深圳 518036)
杨莉丽1,2 王成文2 邹 傲2
高通量测序分析肠易激综合征患者和健康人肠道菌群差异*
1.汕头大学医学院 (广东 汕头 515041)
2.北京大学深圳医院消化内科 (广东 深圳 518036)
杨莉丽1,2王成文2邹 傲2
目的通过高通量测序方法分析肠易激综合征患者(Irritable Bowel Syndrome,IBS)和健康人肠道菌群差异。方法采用Illumima系统MiSeq平台对正常组和IBS组粪便进行16S rRNA测序。结果IBS患者的肠道菌群多样性无显著变化。在门水平上,Firmicutes丰度显著增加(P<0.01),Bacteroidete的丰度和Bacteroidetes/Firmicutes的比值下降。在科水平上,正常组和IBS组粪便肠道菌群中检测到Cryomorphaceae、Erysipelotrichaceae、Campylobacteraceae和Fibrobacteraceae,且丰度变化具有显著差异(P<0.05)。结论IBS患者和健康人相比,肠道菌群多样性未见明显差异。但某些细菌菌种存在显著差异。
肠易激综合征;肠道菌群;高通量测序;16S rRNA
肠易激综合征(Irritable Bowel Syndrome,IBS)是一组以腹痛、腹胀、排便习惯和粪便性状改变为主要临床表现,症状可持续存在或间歇发作,是最常见的肠道功能紊乱性疾病[1]。近年来,许多研究发现IBS患者存在肠道菌群的变化,并认为IBS的发生发展与肠道菌群变化有关。本文通过高通量测序细菌16SrRNA基因序列技术对IBS患者肠道菌群分布进行分析并观察其变化情况。
1 资料和方法
1.1 对象2015年10月至2016年10月在北京大学深圳医院消化科门诊就诊的IBS患者10例,采用罗马Ⅲ诊断标准[2],排除消化道器质性病变。其中男5例,女5例,年龄18~61岁。正常组为10例北京大学深圳医院体检健康者。全部病例无畏寒发热史、胃肠道手术史,无慢性小肠或结肠感染史、慢性肝病、糖尿病、甲亢病史,近3月来未使用过抗生素。
1.2 方法
1.2.1 肠道菌群DNA的提取:使用无菌的离心管收集粪便样本50g,并用肠道菌群细菌基因组提取试剂盒提取制备粪便样本中微生物总DNA,具体方法按说明书进行。
1.2.2 16S rDNA的扩增与测序:用分光光度计对提取的全基因组DNA进行质检,质检合格的细菌进行16S rDNA的V4可变区进行扩增,扩增条件为:94℃变性,30s;50℃退火,30s;72℃延伸,30s。引物为:正向5'-AYTGGGYDTAAAGNG-3',反向5'-TACNVGGGTATCTAATCC-3',PCR产物经均一化处理后建库,PCR体系为25μL,测序平台为Illumima系统MiSeq。
1.3 统计学分析使用SPSS16.0软件对菌群多样性进行t检验,使用mothur软件中metastats命令进行组间统计学差异分析,P<0.05为差异有统计学意义。
2 结 果
2.1 16S rRNA基因测序分析对测序后的结果进行分析,正常组和IBS组的有效序列分别为(67246±19467)和(55564±9892),优化后分别得到(67457±19497)和(55742±9888)条优质序列。在相似度为97%条件下,应用生物信息学软件对序列进行操作单元聚类,共获得47656个OTU代表序列,其中纲水平4763个,科水平47038个,属水平46201个。经绘制Observed species指数稀释曲线、Chao指数稀释曲线,大部分测序的样品接近平台期。分析两组样品的测序结果,发现它们的覆盖率均>97%(P>0.05),表示测序深度已基本覆盖到粪便样本中绝大多数物种,见表1。

表1 正常组和IBS组测得的序列数及覆盖率
表2 FPE作用前后肠道菌科水平变化
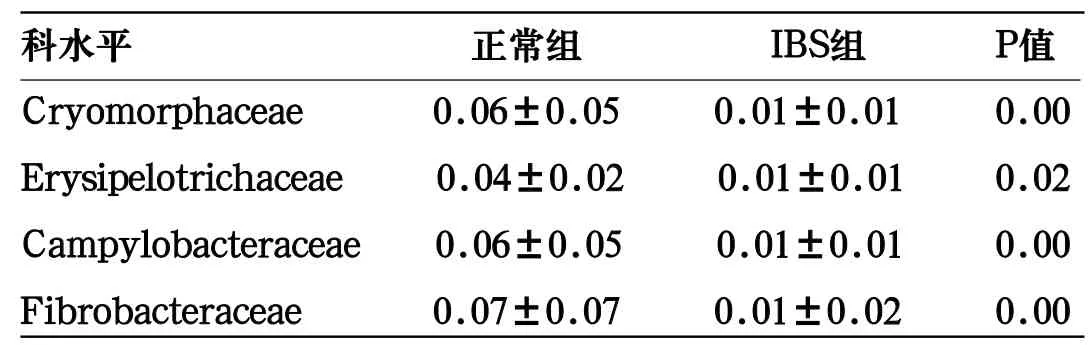
表2 FPE作用前后肠道菌科水平变化
科水平 正常组 IBS组 P值Cryomorphaceae 0.06±0.05 0.01±0.01 0.00 Erysipelotrichaceae 0.04±0.02 0.01±0.01 0.02 Campylobacteraceae 0.06±0.05 0.01±0.01 0.00 Fibrobacteraceae 0.07±0.07 0.01±0.02 0.00
2.2 肠道菌群结构分析对测序序列与16S rDNA数据库进行比对,分析结果发现样本中细菌绝大多数由Firmicutes、Bacteroidetes和Proteobacteria组成,正常组和IBS组Firmicutes序列分别为30467条和39850条,占总菌的比例为67%和73%;Bacteroidetes序列分别为7675条和6203条,占总菌的比例为18%和13%;Proteobacteria序列分别为5462条和6492条,占总菌的比例为11%和13%。统计分析发现与正常组相比,IBS组样品的Firmicutes丰度显著增加(P<0.01),Bacteroidete的丰度和Bacteroidetes/Firmicutes的比值也明显下降(P>0.01),见图1。
对高通量测序数据进一步深入分析,分析结果发现正常组和IBS组肠道菌群主要的科均为Alcaligenaceae、Lachnospiraceae、Rikenellaceae和Ruminococcaceae,分别占总数的8.5%、41.1.1%、8.1%、23.8%和8.1%、42.4%、7.0%、25.1%。统计分析发现正常组和IBS组有4个科Cryomorphaceae、Erysipelotrichaceae、Campylobacteraceae、和Fibrobacteraceae的丰度发生显著变化,具有统计学意义,见表3。
3 讨 论
近年来,国内外大量研究发现IBS与肠道菌群紊乱密切相关,很多研究结果指出肠道菌群失调可能是IBS发病的重要机制。与正常人相比,IBS患者肠道内拟杆菌、双歧杆菌和乳酸杆菌等组成异常,其厚壁门与拟杆菌门比值比正常组高2倍之多[3]。Jalanka-Tuovinen等通过phylogenetic microarray和qPCR实验研究了IBS和正常人的粪便菌群组成,发现患者粪便中的拟杆菌门是正常人的12倍,而对于非培养的梭状芽孢杆菌,正常组则是处理组的35倍[4]。这些研究结果提示IBS患者肠道内菌群与正常人相比具有较大差异。
本研究通过高通量测序方法,取得正常组和IBS患者的粪便样品中微生物的结构和丰度,并分析IBS患者肠道菌种的变化。结果显示,肠道微生物主要由Firmicutes、Bacteroidetes和Proteobacterias构成。在IBS患者中,Firmicutes丰度显著增加(P<0.01),Bacteroidete的丰度和Bacteroidetes/Firmicutes的比值也明显下降(P>0.01)。Bacteroidetes与Firmicutes比值是肠道微生态平衡、肠道健康的重要指标,与国外Rajilic-Stojanovic M等的研究结果一致[5]。此外,对肠道菌群科水平进一步的研究,分析发现正常人组和IBS组在Cryomorphaceae、Erysipelotrichaceae、Campylobacteraceae、和Fibrobacteraceae4个科的丰度变化明显差异,具有统计学意义。Cryomorphaceae属于拟杆菌门,在人体肠道内的丰富程度与高脂饮食、体育锻炼等生活有关[6]。本研究在IBS组中Erysipelotrichaceae明显下降,Erysipelotrichaceae减少可导致IBS的发生[7]。Campylobacteraceae属于变形菌门,与饮食习惯和身体营养状况有关[8],Fibrobacteraceae常在等多种动物的肠道中检出,如食草类动物:牛、羊,Fibrobacteraceae与内脏神经高敏感性有关[9]。本研究结果提示IBS患者和健康人相比,菌群多样性未见明显差异。但某些细菌菌种存在显著差异。Campylobacteraceae、Cryomorphaceae、Erysipelotrichaceae和Fibrobacteraceae这4科,可能在IBS的诊断和治疗中具有积极作用。
因本研究样本量相对较少,故未对IBS亚型进行分组研究,IBS亚型对肠道微生物也有一定影响,因此肠道菌群对IBS发病影响的具体机制仍需进一步研究。
[1] DrossmanDA.The functional gastrointestinal disorders and the Rome III process[J].Gastroenterology,2006,130(5)∶377-390.
[2] Drossman DA,Corrazziari E,Delvaux M,et a1.Rome Ⅲ∶The functional gastrointestinal disorders[J].Gastroenterology,2006,130(130)∶1466-1479.
[3] Dai, C.Z., C. Q.Jiang, M.Ma, X. Y.Jiang, L. J., Probiotics and irritable bowel syndrome[J].World J Gastroenterol,2013,19(36)∶5973-5980.
[4] Jalanka-Tuovinen J, S.J., Salonen A, Immonen O, Garsed K,Kelly FM, Zaitoun A, Palva A, Spiller RC, de Vos WM., Faecal microbiota composition and host-microbe cross-talk following gastroenteritis and in postinfectious irritable bowel syndrome[J].Gut,2013∶ p. Epub ahead of print.
[5] Rajilic-Stojanovic M, Biagi E, HeiligHG,etal.Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome[J].Gastroenterology.2011,141(5)∶1792-1801.
[6] Dai C, Zheng CQ, Jiang M, et al.Probiotics and irritable bowel syndrome[J].World J Gastroenterol,2013,19(36)∶5973-5980.
[7] 陈宏运,崔红燕,吴彬彬,等.植物发酵液对D-半乳糖致衰老模型小鼠的抗氧化活性研究[J].现代食品科技,2015,31(8)∶7-11,17.
[8] Roberts LM, McCahon D, Holder R, et al.Arandomised controlled trial of a probiotic 'functional food' in the management of irritable bowel syndrome[J].BMC Gastroenterol,2013,13(1)∶45
[9] Zhou XY, Li M, Li X, et al.Visceral hypersensitive rats share common dysbiosis features with irritable bowel syndrome patients[J].World J Gastroenterol,2016,22(22)∶5211-5227.
High-throughput Sequencing Analysis of Intestinal Microbiota in Patients with Irritable Bowel Syndrome and Healthy Individuals*
YANG Li-li, WANG Cheng-wen,ZOU Ao. Shantou University Medical College, Shantou 515041, Guangdong Province, China
ObjectiveTo compare differences of intestinal microbiotabetween IBS patients andhealthy by using high-throughput sequencing technology.MethodsThe feces were collected IBS group andhealthy group. The community structure and abundance of intestinal microbiota were examined via analyzing 16S rRNA high throughput sequencing using Illumima System.ResultsThe diversities of intestinal bacteria were no significant differences in IBS group. In phyla level, the abundance of Firmicutes was significantlyincreased (P<0.01), while the abundance of Bacteroidetesand the ratio ofBacteroidetes/Firmicutes were decreased (P<0.01). Besides, in family level, cryomorphaceae,erysipelotrichac eae. Campylobacteraceae and fibrobacteraceae were detected in IBS group and healthy group.Their abundance were significantly difference, respectively (P<0.05).ConclusionNo significantdifferences in the bacterial diversity indices are found, but some bacterial species significantly differbetween the patients with IBS and healthy controls.
Irritable Bowel Syndrome; Intestinal Microbiota; High-throughput Sequencing; 16S rRNA
R378.2
A
深圳市科技计划项目:No.JCYJ201404151625429
10.3969/j.issn.1009-3257.2017.05.015
杨莉丽,女,消化内科专业,副主任医生,主要研究方向:胃肠功能性疾病
王成文
2017-09-21
猜你喜欢
中老年保健(2022年2期)2022-08-24
中国饲料(2022年5期)2022-04-26
昆明医科大学学报(2022年1期)2022-02-28
疯狂英语·新悦读(2021年10期)2021-11-23
科学(2020年4期)2020-11-26
透析与人工器官(2020年1期)2020-11-16
铁道通信信号(2019年8期)2019-10-10
中国发展观察(2017年8期)2017-04-26
中国当代医药(2015年33期)2015-03-01
现代检验医学杂志(2015年4期)2015-02-06
