
米拉贝隆有关物质的合成
2017-10-14 02:27鲍雨贤张庆华徐广宇
湖南师范大学自然科学学报 2017年5期
鲍雨贤,张 为,陈 波,张庆华,徐广宇
(1.湖南师范大学化学化工学院,中国 长沙 410081;2.湖南师范大学医学院,中国 长沙 410013;3.湖南方盛制药股份有限公司,中国 长沙 410205)
米拉贝隆有关物质的合成
鲍雨贤1,张 为2,陈 波3,张庆华3,徐广宇1
(1.湖南师范大学化学化工学院,中国 长沙 410081;2.湖南师范大学医学院,中国 长沙 410013;3.湖南方盛制药股份有限公司,中国 长沙 410205)
为加强对米拉贝隆原料药的质量控制,以硝基苯乙腈和扁桃酸为原料合成了米拉贝隆的8个有关物质A~H,所合成的化合物结构经1H-NMR,13C-NMR与MS 谱确证,其纯度经HPLC 法检测,表明有关物质A~H可作为米拉贝隆质量控制的杂质对照品.
米拉贝隆;工艺;杂质;合成
AbstractTo strengthen the quality control of mirabegron API, eight related substances of mirabegron were synthesized from nitrophenylacetonitrile and mandelic acid. The structures of impuritieA~Hwere confirmed by1H,13C NMR and MS, and their purities were determined by HPLC analysis. The impurities synthesized could be used as the reference substances of the related substances in the quality control of mirabegron.
Keywordsmirabegron; process; related substances; synthesis
米拉贝隆(Mirabegron)由日本安斯泰来制药公司开发,于2011年在日本上市,2012年美国食品药品监督管理局批准用于治疗成年人膀胱过度活动症[1].米拉贝隆可以选择性地与膀胱肌肉的β3-肾上腺能受体相结合,将其激活而松弛膀胱肌肉、提高膀胱的容量,利于储尿,可以减少尿频、遗尿、尿急等症状,有良好的疗效和耐受性,不良反应少[2-3].药物不良反应是指用药过程中出现的与治疗作用无关的不良结果,它与药品自身的药理活性相关外,亦与其含有的杂质有关,将杂质控制在一个安全合理的范围内,关系到药品的质量和安全[4].因此制备米拉贝隆的杂质对照品,用于米拉贝隆的原料药生产过程中的质量控制研究具有重要实用价值.
1 米拉贝隆的制备方法
米拉贝隆的制备方法有很多相关的文献报道[5-13],适合工业化生产的合成路线如图1[9]:
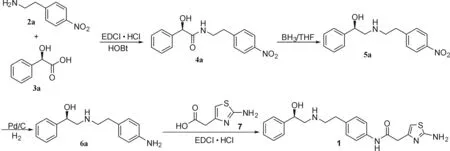
图1 米拉贝隆的合成Fig.1 Synthesis of mirabegron
该路线以4-硝基苯乙胺和D-扁桃酸为原料,首先缩合得到酰胺4a,再经硼烷还原酰胺得化合物5a,然后5a用钯碳加氢还原得化合物6a,最后6a与2-氨基噻唑-4-乙酸7缩合得到米拉贝隆.笔者对此合成路线进行分析并定向合成了工艺过程中产生的8个有关物质Imp.A~H(图2).
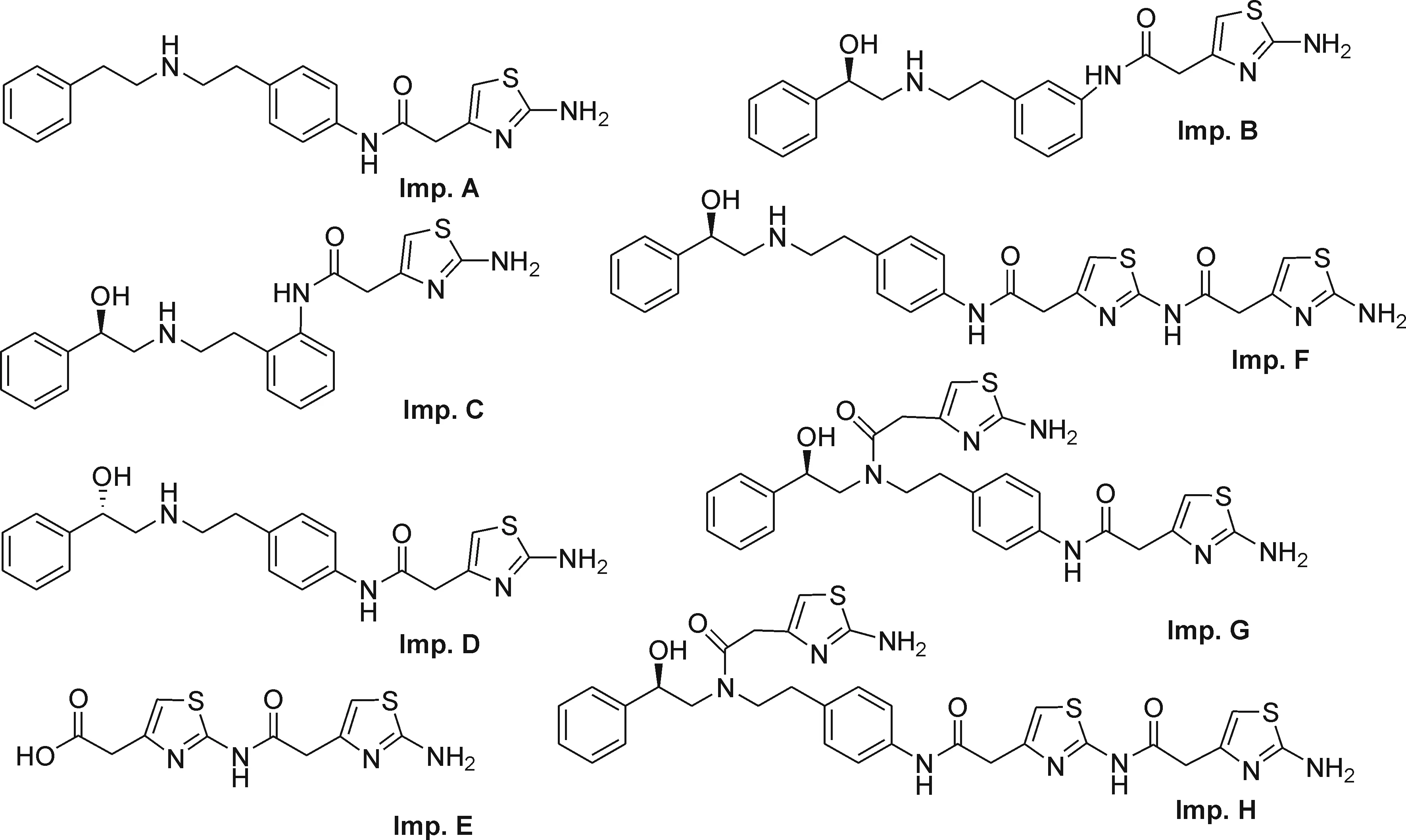
图2 米拉贝隆有关物质Fig.2 The related substances of mirabegron
2 实验部分
2.1 仪器与试剂
高效液相色谱仪(岛津SPD-15C),超导核磁共振仪(Brücker Avance-500),质谱仪(Waters Xevo).用于合成的试剂均为市售分析纯.HPLC条件,色谱柱:C18柱 (4.6 mm×250 mm, 5 μm);流动相A:V(0.01 mol/L磷酸氢二钾溶液)∶V(0.01 mol/L磷酸二氢钾溶液)=1∶1;流动相B:乙腈;梯度洗脱(0 min,B 20%;10 min,B 50%;15 min,B 80%;30 min,B 20%);流速:0.8 mL/min;检测波长210 nm;柱温40 ℃.
2.2 2-(2-氨基噻唑-4-基)-N-4-(2-(苯乙基氨基)乙基)苯乙酰胺(Imp.A)的合成
如图3,将(R)-2-((4-硝基苯乙基)氨基)-1-苯基乙醇盐酸盐5a[9](2.00 g,6.2 mmol),0.4 g Pd/C(10%,含水68.14%),浓硫酸(0.74 g,7.4 mmol)与40 mL冰乙酸混合,常压通入氢气,加热至38 ℃反应5 h.原料反应完毕后过滤,滤液减压抽除溶剂,加入20% 碳酸钠溶液调节溶液pH至9,乙酸乙酯(3×20 mL)萃取,合并有机相,30 mL饱和食盐水洗涤,无水硫酸钠干燥,减压抽除溶剂,粗品用甲醇-异丙醚重结晶得1.24 g化合物8,黄色固体,收率83.3%.1H NMR (500 MHz, DMSO-d6):δ9.17 (brs, 2H), 7.35~7.32 (m, 2H), 7.27~7.24 (m, 3H), 6.95 (d,J=8.0 Hz, 2H), 6.63 (d,J=8.0 Hz, 2H), 5.99 (brs, 2H), 3.13 (s, 2H), 3.05 (s, 2H), 3.00~2.96 (m, 2H), 2.84~2.80 (m, 2H);1H NMR (500 MHz, CD3OD):δ7.36~7.25 (m, 5H), 7.07 (d,J=8.0 Hz, 2H), 6.79 (d,J=8.0 Hz, 2H), 3.27~3.19 (m, 4H), 3.01~2.88 (m, 4H); ESI-MS(m/z): 241.2[M+H]+.
将化合物8(1.24 g,5.2 mmol),2-氨基噻唑-4-乙酸7(1.06 g,6.7 mmol),1-(3-二甲胺基丙基)-3-乙基碳二亚胺盐酸盐(EDCI·HCl)(1.29 g,6.7 mmol)和浓盐酸(0.66 g,6.7 mmol)溶于19 mL水中,在15 ℃下搅拌反应3 h.原料反应完毕后,缓慢滴加2 mol/L氢氧化钠溶液调节pH至9,过滤,粗品用乙醇-水重结晶,得到1.76 g杂质A,白色固体,收率89.3%.纯度99.01%(相对1的保留时间为1.17).1H NMR (500 MHz, CD3OD):δ7.56 (d,J=8.0 Hz, 2H), 7.35~7.32 (m, 2H), 7.28~7.22 (m, 5H), 6.36 (s, 1H), 3.56 (s, 2H), 3.27~3.23 (m, 4H), 3.01~2.95 (m, 4H);13C NMR (125 MHz, CD3OD):δ170.23, 169.36, 144.39, 137.58, 136.34, 132.04, 128.78, 128.62, 128.37, 126.91, 120.34, 103.72, 48.71, 48.67, 38.86, 31.98, 31.41; ESI-MS (m/z): 381.3[M+H]+.
2.3 (R)-2-(2-氨基噻唑-4-基)-N-3-(2-((2-羟基-2-苯乙基)氨基)乙基)苯乙酰胺(Imp.B)的合成
如图4,将3-硝基苯乙腈9a(1.50 g,9.3 mmol)置于100 mL圆底烧瓶中,加入30 mL THF,冰盐浴下搅拌降温至-5 ℃以下,滴加1 mol/L BH3的THF溶液(20.5 mL,20.5 mmol),滴加完毕后升温至回流反应5 h.原料反应完后,缓慢滴加16.09 g 3%盐酸-甲醇溶液,滴完加热回流1 h.40 ℃下减压抽除溶剂,残余物加入7.5 mL异丙醇,室温搅拌0.5 h,过滤,少量异丙醇洗涤,干燥,得到1.30 g 3-硝基苯乙胺盐酸盐2b,白色固体,收率69.5%.1H NMR (500 MHz, D2O):δ8.18~8.14 (m, 2H), 7.71 (d,J=7.5 Hz, 1H), 7.59 (t,J=8.0 Hz, 1H), 3.33 (t,J=7.5 Hz, 2H), 3.12 (t,J=7.5 Hz, 2H);13C NMR (125 MHz, D2O):δ148.05, 138.44, 135.70, 130.01, 123.67, 122.38, 40.04, 32.31.

图4 杂质B的合成Fig.4 The synthesis of impurity B
在25 mL圆底烧瓶中加入化合物2b(1.30 g,6.4 mmol),D-扁桃酸3a(0.98 g,6.4 mmol),1-羟基苯并三氮唑(0.87 g,6.4 mmol),EDCI·HCl(1.23 g,6.4 mmol),三乙胺(0.65 g,6.4 mmol)和6.5 mL DMF,在40 ℃下加热搅拌5 h.原料反应完毕后,加30 mL水稀释,乙酸乙酯(3×20 mL)萃取.有机相依次用30 mL 2 mol/L盐酸、饱和Na2CO3溶液、饱和氯化钠溶液洗涤,无水硫酸钠干燥.40 ℃下减压抽除溶剂,得淡黄色油状物,粗品用甲苯重结晶得到1.37 g化合物4b,白色固体,收率71.1%.1H NMR (500 MHz, CDCl3):δ8.06 (d,J=8.0 Hz, 1H), 7.97 (s, 1H), 7.42~7.31 (m, 7H), 6.21 (brs, 1H), 5.00 (s, 1H), 3.64~3.51 (m, 2H), 2.89 (td,J=7.0, 1.5 Hz, 2H).
将化合物4b(1.37 g,4.6 mmol)置于100 mL圆底烧瓶中,加入25 mL THF,冰盐浴下搅拌并降温至-5 ℃以下,滴加1 mol/L BH3的THF溶液(10.0 mL,10.0 mmol),滴加完毕后升温至回流反应5 h.原料反应完后,缓慢滴加14.24 g 4%盐酸-甲醇溶液,滴完加热回流1 h.40 ℃下减压抽除溶剂,加入7.0 mL异丙醇,室温搅拌析晶,过滤,少量异丙醇洗涤,干燥,得到1.15 g化合物5b,白色固体,收率78.1%.1H NMR (500 MHz, DMSO-d6):δ9.52 (brs, 1H), 9.02 (brs, 1H), 8.17 (t,J=2.0 Hz, 1H), 8.13 (dd,J=8.5, 1.5 Hz, 1H), 7.76 (d,J=8.0 Hz, 1H), 7.65 (t,J=8.0 Hz, 1H), 7.41~7.37 (m, 4H), 7.33~7.30 (m, 1H), 6.23 (brs, 1H), 5.03 (d,J=9.5 Hz, 1H), 3.27~3.26 (m, 2H), 3.21~3.17 (m, 3H), 3.06~3.00 (m, 1H).
在25 mL三口烧瓶中,加入化合物5b(1.15 g,3.6 mmol),0.06 g Pd/C(10%,含水68.14%)和10 mL甲醇.抽真空,氢气置换3次.常压通入氢气,室温搅拌10 h.原料反应完毕后过滤,少量甲醇洗涤.50 ℃减压抽除溶剂,得白色油状物.乙酸乙酯重结晶,干燥,得到1.03 g化合物6b,白色固体,收率98.7%.1H NMR (500 MHz, DMSO-d6):δ9.17 (brs, 1H), 8.89 (brs, 1H), 7.40~7.37 (m, 4H), 7.33~7.30 (m, 1H), 6.95 (t,J=7.5 Hz, 1H), 6.44~6.41 (m, 2H), 6.36 (d,J=7.5 Hz, 1H), 6.20 (d,J=4.0 Hz, 1H), 5.08 (brs, 2H), 4.98 (dt,J=10.5, 3.0 Hz, 1H), 3.17~3.09 (m, 3H), 3.04~2.99 (m, 1H), 2.89~2.78 (m, 2H).
将化合物6b(1.03 g,3.5 mmol),2-氨基噻唑-4-乙酸7(0.72 g,4.6 mmol),浓盐酸(0.45 g,4.6 mmol),EDCI·HCl(0.88 g,4.6 mmol)和10 mL水混合,室温搅拌3 h.原料反应完毕后滴加2 mol/L氢氧化钠溶液调节pH至9,滴完室温搅拌1 h.加10 mL水稀释,二氯甲烷萃取(3×20 mL), 40 ℃下减压抽除溶剂,以V(乙酸乙酯)/V(石油醚)=3/1为洗脱剂经硅胶柱层析分离得到1.08 g杂质B,淡黄色固体,收率77.4%.纯度97.62%(相对1的保留时间为1.10).1H NMR (500 MHz, CD3OD):δ7.45~7.41 (m, 2H), 7.33~7.28 (m, 4H), 7.25~7.20 (m, 2H), 6.95 (d,J=7.5 Hz, 1H), 6.36 (s, 1H), 4.74 (dd,J=8.0, 5.0 Hz, 1H), 3.56 (s, 2H), 2.93~2.86 (m, 2H), 2.82~2.77 (m, 4H);13C NMR (125 MHz, CD3OD):δ170.22, 169.29, 144.51, 142.99, 140.15, 138.60, 128.61, 128.02, 127.20, 125.57, 124.22, 120.00, 117.83, 103.61, 71.79, 56.42, 50.11, 38.98, 35.20; ESI-MS (m/z): 397.3[M+H]+.
2.4 (R)-2-(2-氨基噻唑-4-基)-N-2-(2-((2-羟基-2-苯乙基)氨基)乙基)苯乙酰胺(Imp.C)的合成
如图5,以2-硝基苯乙腈9b为原料(1.50 g,9.3 mmol),参照杂质B合成路线的方法,合成得到1.42 g杂质C,淡黄色固体,总收率38.7%.纯度99.76%(相对1的保留时间为1.12).1H NMR (500 MHz, CD3OD):δ7.57 (d,J=7.5 Hz, 1H), 7.34~7.29 (m, 4H), 7.25~7.19 (m, 3H), 7.13 (t,J=7.5 Hz, 1H), 6.37 (s, 1H), 4.75 (dd,J=8.5, 4.0 Hz, 1H), 3.59 (s, 2H), 2.86~2.73 (m, 6H);13C NMR (125 MHz, CD3OD):δ170.43, 169.92, 144.49, 143.12, 135.62, 133.39, 129.90, 128.02, 127.17, 126.63, 125.78, 125.64, 125.09, 103.79, 71.87, 56.61, 49.27, 38.75, 31.51; ESI-MS (m/z): 397.3[M+H]+.

图5 杂质C的合成Fig.5 The synthesis of impurity C
2.5 (S)-2-(2-氨基噻唑-4-基)-N-4-(2-((2-羟基-2-苯乙基)氨基)乙基)苯乙酰胺(Imp.D)的合成
如图6,以4-硝基苯乙胺盐酸盐2a(1.50 g,9.9 mmol) 和L-扁桃酸3b(2.00 g,9.9 mmol)为原料,参照杂质B合成路线的方法合成得到2.58 g杂质D,淡黄色固体,总收率65.9%,纯度99.32%(相对1的保留时间为1.00).1H NMR (500 MHz, CD3OD):δ7.48 (d,J=8.5 Hz, 2H), 7.32~7.28 (m, 4H), 7.24~7.21 (m, 1H), 7.14 (d,J=8.5 Hz, 2H), 6.35 (s, 1H), 4.73 (dd,J=8.0, 4.5 Hz, 1H), 3.55 (s, 2H), 2.88~2.73 (m, 6H);13C NMR (125 MHz, CD3OD):δ170.21, 169.20, 144.55, 143.06, 136.68, 135.35, 128.63, 128.03, 127.20, 125.58, 120.05, 103.62, 71.90, 56.50, 50.29, 38.92, 34.70; ESI-MS(m/z): 397.3[M+H]+.

图6 杂质D的合成Fig.6 The synthesis of impurity D
2.6 2-(2-(2-(2-氨基噻唑-4-基)乙酰氨基)噻唑-4-基)乙酸(Imp.E)的合成
如图7,将硫脲(20.0 g,262.7 mmol)溶于100 mL水中,0~5 ℃下滴加4-氯乙酰乙酸乙酯(44.5 g,270.1 mmol),滴完继续搅拌5 h.原料反应完毕后,滴加浓氨水调节pH至9,搅拌0.5 h后过滤,滤饼用10 mL水洗,干燥得42.92 g化合物10,淡黄色固体,收率87.7%.1H NMR (500 MHz, CDCl3):δ6.34 (s, 1H), 5.33 (brs, 2H), 4.18 (q,J=7.0 Hz, 2H), 3.56 (s, 2H), 1.27 (t,J=7.0 Hz, 3H).

图7 杂质E的合成Fig.7 The synthesis of impurity E
将化合物10(20.00 g,107.4 mmol)和4-二甲氨基吡啶(4.00 g,32.7 mmol)溶于二氯甲烷100 mL,室温搅拌下滴加二碳酸二叔丁酯(28.13 g,128.9 mmol),滴完室温搅拌.反应完毕后依次用30 mL 1 mol/L 盐酸、饱和食盐水洗涤,有机相用无水硫酸钠干燥,减压抽除溶剂,所得残余物和LiOH一水物(9.01 g,214.8 mmol)、乙醇50 mL及水25 mL混合,40 ℃下搅拌2 h.反应完毕后,加60 mL水稀释,冰浴下滴加冰醋酸调节pH至5~6,继续搅拌1 h后过滤,5 mL水洗,干燥,得到21.81 g化合物11,白色固体,收率88.4%.1H NMR (500 MHz, DMSO-d6):δ12.22 (brs, 1H), 11.47 (brs, 1H), 6.88 (s, 1H), 3.54 (s, 2H), 1.47 (s, 9H).
将化合物11(10.40 g,40.3 mmol),2-氨基噻唑-4-乙酸乙酯10(5.00 g,26.9 mmol)和EDCI·HCl(6.18 g,32.2 mmol)溶于20 mL DMF中,于50 ℃加热搅拌48 h.反应完毕后加60 mL水稀释,乙酸乙酯萃取(3×30 mL),有机相依次用 20 mL 1 mol/L 盐酸、饱和食盐水洗涤,无水硫酸钠干燥,减压抽除溶剂,得到8.73 g化合物12,黄色固体,收率76.2%.1H NMR (500 MHz, CDCl3):δ11.56 (brs, 1H), 10.55 (brs, 1H), 6.80 (d,J=7.0 Hz, 2H), 4.13 (q,J=7.0 Hz, 2H), 3.83 (s, 2H), 3.69 (s, 2H), 1.56 (s, 9H), 1.21 (t,J=7.0 Hz, 3H).
取化合物12(8.70 g,20.4 mmol),加入LiOH一水物(2.23 g,53.0 mmol)、乙醇20 mL和水10 mL,于40 ℃下加热搅拌2 h.反应完毕后加30 mL水稀释,冰浴下滴加冰醋酸调节pH至5~6,继续搅拌1 h后过滤,5 mL水洗,干燥,得到7.14 g化合物13,淡黄色固体,收率87.9%.1H NMR (500 MHz, DMSO-d6):δ12.28 (brs, 1H), 11.42 (brs, 1H), 6.92 (d,J=4.0 Hz, 2H), 3.74 (s, 2H), 3.58 (s, 2H), 1.45 (s, 9H).
在25mL圆底烧瓶中,加入化合物13(1.22 g,3.5 mmol)与3 mL三氟乙酸,40 ℃下搅拌2 h后减压抽除溶剂,加入 5 mL 饱和碳酸钠溶液,室温搅拌0.5 h,过滤,滤液用1 mol/L盐酸调节pH至5,析出固体,过滤,少量水洗滤饼,干燥得0.89 g杂质E,淡黄色固体,收率97.4%.纯度95.68%(相对1的保留时间为0.18).1H NMR (500 MHz, DMSO-d6):δ12.38 (brs, 1H), 8.64 (brs, 2H), 6.97 (s, 1H), 6.65 (s, 1H), 3.76 (s, 2H), 3.60 (s, 2H);13C NMR (125 MHz, DMSO-d6):δ172.01, 170.18, 167.19, 159.23, 157.65, 144.81, 110.79, 105.53, 37.29, 35.36; ESI-MS (m/z): 299.0[M+H]+.
2.7(R)-2-(2-氨基噻唑-4-基)-N-((4-(2-(4-(2-((2-羟基-2-苯乙基)氨基)乙基)苯基)氨基)-2-氧代乙基)噻唑-2-基)乙酰胺(Imp.F)的合成
如图8,在25 mL圆底烧瓶中,加入杂质E(65 mg,0.16 mmol)、化合物6a[9](60 mg,0.23 mmol)、EDCI·HCl (60 mg,0.3 mmol)和10 mL水,用浓盐酸调节溶液pH至3,室温搅拌3 h后补加EDCI·HCl(60 mg,0.3 mmol),室温搅拌过夜,滴加2 mol/L 氢氧化钠溶液调节pH至9,继续搅拌0.5 h后过滤,少量水洗,干燥,粗品以V(乙酸乙酯)/V(石油醚)/V(甲醇)=20/10/1的洗脱剂经硅胶柱层析分离得到78.0 mg 杂质F,淡黄色固体,收率62.1%.纯度95.21%(相对1的保留时间为1.22).1H NMR (500 MHz, CD3OD):δ7.40 (d,J=8.5 Hz, 2H), 7.24~7.19 (m, 4H), 7.14~7.12 (m, 1H), 7.05 (d,J=8.0 Hz, 2H), 6.82 (s, 1H), 6.28 (s, 1H), 4.68 (t,J=6.5 Hz, 1H), 3.64 (s, 2H), 3.56 (s, 2H), 2.86~2.68 (m, 6H);13C NMR (125 MHz, CD3OD):δ170.35, 169.28, 168.71, 158.36, 144.61, 143.31, 142.71, 136.77, 134.96, 128.66, 128.08, 127.31, 125.60, 120.12, 110.37, 104.20, 71.42, 56.07, 50.03, 38.95, 37.65, 34.18; ESI-MS(m/z): 537.2[M+H]+.
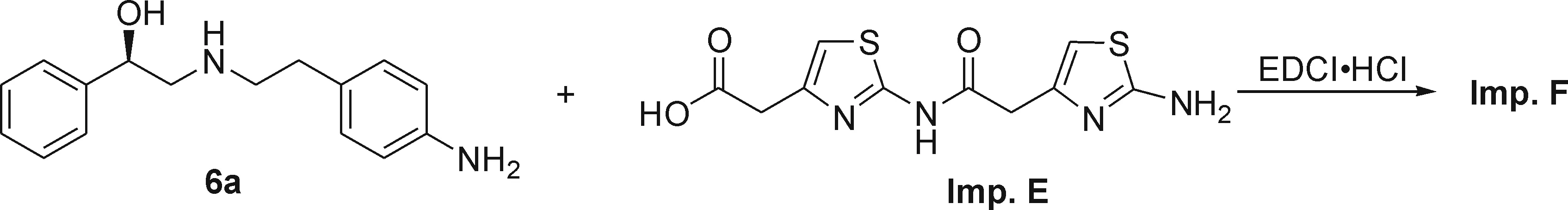
图8 杂质F的合成Fig.8 The synthesis of impurity F
2.8(R)-2-(2-氨基噻唑-4-基)-N-(4-(2-(2-氨基噻唑-4-基)乙酰氨基)苯乙基)-N-(2-羟基-2-苯乙基)乙酰胺(Imp.G)的合成
如图9,在50 mL圆底烧瓶中加入化合物11(8.80 g,34.1 mmol)和20 mL二氯甲烷,冰盐浴下滴加7.86 g草酰氯(62.0 mmol),滴完继续搅拌1 h,减压抽除溶剂,所得淡黄色固体加入10 mL二氯甲烷溶解待用.将化合物5a(10.00 g,31.0 mmol)、碳酸钾(8.56 g,62.0 mmol)和20 mL二氯甲烷混合,冰盐浴下滴加上述酰氯的二氯甲烷溶液,滴完继续在室温下搅拌3 h,加10 mL水稀释,分液,有机相用水洗,无水硫酸钠干燥,减压抽除溶剂,得16.16 g化合物14,黄色固体,收率99.1%.1H NMR (500 MHz, DMSO-d6):δ11.36 (s, 1H), 8.16 (d,J=8.5 Hz, 1H), 8.09 (d,J=8.5 Hz, 1H), 7.50 (d,J=9.0 Hz, 1H), 7.44 (d,J=8.5 Hz, 1H), 7.38~7.28 (m, 5H), 6.76 (s, 0.35H), 6.67 (s, 0.55H), 5.67 (s, 0.65H), 5.46 (s, 0.36H), 4.81~4.76 (m, 1H), 3.72~3.51 (m, 6H), 2.97~2.88 (m, 2H), 1.47 (s, 9H).
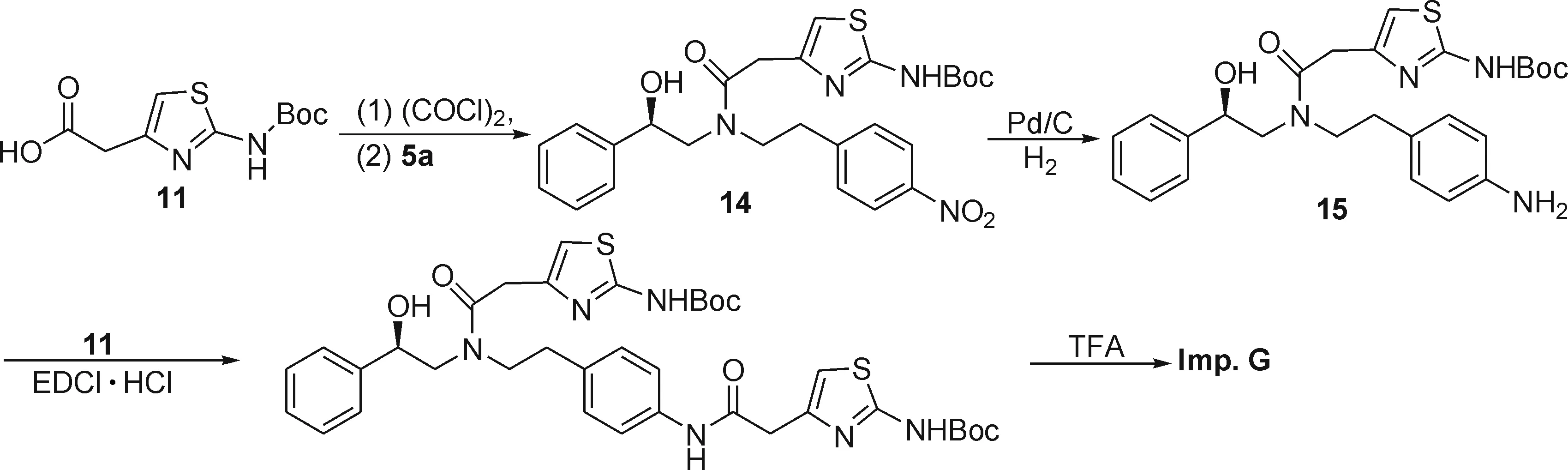
图9 杂质G的合成Fig.9 The synthesis of impurity G
在250 mL三口烧瓶中加入化合物14(10.00 g,19.0 mmol),3.00 g Pd/C(10%,含水68.14%)和150 mL甲醇,减压抽除空气,常压通入氢气,室温搅拌过夜,过滤,减压抽除溶剂后得到9.25 g化合物15,黄色固体,收率98.1%.1H NMR (500 MHz, DMSO-d6):δ11.40 (s, 1H), 7.45~7.22 (m, 7H), 6.82 (dd,J=15.5, 8.0 Hz, 2H), 6.70 (s, 0.62H), 6.63 (s, 0.38H), 6.49 (t,J=6.5 Hz, 2H), 5.67 (s, 0.48H), 5.45 (s, 0.53H), 4.81~4.78 (m, 1H), 3.72~3.20 (m, 6H), 2.62~2.52 (m, 2H), 1.47 (s, 9H).
将化合物15(1.70 g,3.4 mmol)、化合物11(1.33 g,5.1 mmol)和EDCI·HCl(0.79 g,4.1 mmol)溶于20 mL DMF中,于50 ℃加热搅拌48 h,反应完毕后加50 mL水稀释,乙酸乙酯萃取(3×30 mL),有机相依次用 20 mL 1 mol/L 盐酸、饱和食盐水洗涤,无水硫酸钠干燥,减压抽除溶剂,往残余物中加入3 mL三氟乙酸,于40 ℃下搅拌2 h后减压抽除溶剂,加入10 mL 饱和碳酸钠与30 mL 乙酸乙酯,室温搅拌0.5 h,乙酸乙酯萃取(2×30 mL),20 mL水洗,有机相用无水硫酸钠干燥,减压抽除溶剂,残余物以V(乙酸乙酯)/V(石油醚)/V(甲醇)=20/10/1的洗脱剂经硅胶柱层析分离得1.02 g杂质G,淡黄色固体,收率57.3%,纯度95.1%(相对1的保留时间为0.95).1H NMR (500 MHz, CD3OD):δ7.48 (dd,J=16.5, 8.0 Hz, 2H), 7.37~7.30 (m, 4H), 7.29~7.24 (m, 1H), 7.12 (dd,J=8.0, 6.5 Hz, 2H), 6.35 (s, 1H), 6.16 (s, 0.53H), 6.10 (s, 0.45H), 4.89 (ddd,J=12.5, 8.0, 4.5 Hz, 1H), 3.73~3.23 (m, 8H), 2.84~2.74 (m, 2H);13C NMR (125 MHz, CD3OD):δ171.81, 170.21, 170.04, 169.21, 144.74, 142.74, 141.32, 136.76, 135.18, 129.04, 127.99, 127.43, 125.77, 120.08, 103.63, 103.19, 71.32, 56.07, 51.39, 38.93, 36.15, 29.27; ESI-MS (m/z): 537.3[M+H]+.
2.9(R)-2-(2-氨基噻唑-4-基)-N-(4-(2-(2-(2-(2-氨基噻唑-4-基)乙酰氨基)噻唑-4-基)乙酰氨基)苯乙基)-N-(2-羟基-2-苯乙基)乙酰胺(Imp.H)的合成
如图10,将化合物15(1.00 g,2.0 mmol)、化合物13(1.20 g,3.0 mmol)和EDCI·HCl(0.46 g,2.4 mmol)溶于20 mL DMF中,于50 ℃加热搅拌48 h,反应完毕后加50 mL水稀释,乙酸乙酯萃取(3×30 mL),有机相依次用20 mL 1 mol/L盐酸、饱和食盐水洗涤,无水硫酸钠干燥,减压抽除溶剂,往残余物中加入3 mL三氟乙酸,40 ℃下搅拌2 h后减压抽除溶剂,往残余物加入10 mL 20%碳酸钠溶液与30 mL乙酸乙酯,室温搅拌0.5 h,乙酸乙酯萃取(2×30 mL),20 mL水洗,有机相用无水硫酸钠干燥,减压抽除溶剂,残余物以V(乙酸乙酯)/V(石油醚)/V(甲醇)=20/10/1的洗脱剂经硅胶柱层析分离得0.83 g杂质H,淡黄色固体,收率60.8%.纯度95.9%(相对1的保留时间为1.19).1H NMR (500 MHz, CD3OD):δ7.48 (dd,J=17.0, 8.5 Hz, 2H), 7.37~7.24 (m, 5H), 7.16~7.10 (m, 2H), 6.91 (s, 1H), 6.38 (s, 1H), 6.14 (s, 0.44H), 6.10 (s, 0.36H), 4.89 (ddd,J=12.5, 7.5, 4.5 Hz, 1H), 3.72 (s, 2H), 3.66 (s, 2H), 3.60~3.23 (m, 6H), 2.90~2.74 (m, 2H);13C NMR (125 MHz, CD3OD):δ171.80, 170.33, 170.04, 169.25, 168.65, 158.31, 144.71, 143.32, 142.66, 142.30, 137.02, 134.36, 129.06, 128.18, 127.25, 125.79, 120.12, 110.37, 104.20, 103.21, 71.34, 56.02, 51.28, 38.98, 37.63, 35.99, 32.47; ESI-MS (m/z): 677.2[M+H]+.

图10 杂质H的合成Fig.10 The synthesis of impurity H
3 结果与讨论
杂质A~H除A已有文献[14-15]报道外,其余7个杂质都为新合成.文献报道杂质A由苯乙酸或苯乙酰氯为原料和4-硝基苯乙胺反应后,再经硼烷还原、催化氢化,最后与2-氨基噻唑-4-乙酸缩合经4步反应合成得到,步骤长、收率低、成本高、操作复杂.笔者利用米拉贝隆合成工艺中的中间体5a为原料经两步反应即可合成得到杂质A,路线简洁,收率高.杂质A是在还原酰胺化合物4a和还原硝基化合物5a时生成的脱羟基副产物随后续反应带入.笔者发现通过对产品进行重结晶纯化不能去除杂质A,但可以对6a的合成反应条件(如反应温度和溶液的pH值)进行控制以有效地降低脱羟基副产物产生.杂质B与杂质C由起始原料4-硝基苯乙胺中的间、邻位硝基苯乙胺杂质随后续合成反应得到.可以通过对终产品进行重结晶去除.杂质D是原料D-扁桃酸中L-异构体随后续反应产生的对映异构体杂质,可以选择合适的供应商将杂质降低到药典规定检测限以下.杂质E是最后一步反应两分子的2-氨基噻唑-4-乙酸缩合产生,杂质F由米拉贝隆中芳伯胺基与2-氨基噻唑-4-乙酸缩合产生,杂质G是米拉贝隆中脂肪仲胺基与2-氨基噻唑-4-乙酸缩合产生的,杂质H是米拉贝隆与两分子2-氨基噻唑-4-乙酸缩合产生的.对最后一步缩合反应的条件如投料比例和反应温度等进行控制即可降低杂质E~H的含量.
本文合成了米拉贝隆的8个有关物质并进行了结构表征.杂质的来源及去除方法在本文中也进行了简要的讨论.本研究可为米拉贝隆的研制与生产提供杂质对照品的技术方法,对于系统建立米拉贝隆原料药及其制剂的有关物质分析控制具有重要的参考价值.
[1] YAO S. FDA approves myrbetriq for overactive bladder[EB/OL].[2012-06-28]. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm310096.htm.
[2] KHULLAR V, AMARENCE G, ANGULO J C,etal. Efficacy and tolerability of mirabegron, aβ3-adrenoceptor agonist, in patients with overactive bladder: results from a randomised european-australian phase 3 trial[J]. Eur Urol, 2013,63(2):283-295.
[3] KRAUWINKEL W, DIJK J V, SCHADDELEE M,etal. Pharmacokinetic properties of mirabegron, aβ3-adrenoceptor agonist: results from two phase I, randomized, multiple-dose studies in healthy young and elderly men and women[J]. Clin Ther, 2012,34(10):2144-2160.
[4] 国家食品药品监督管理总局. 化学药物杂质研究的技术指导原则[Z]. 2005-03-18.
[5] MARUYAMA T, SUZUKI T, ONDA K,etal. Amide derivatives or salts thereof: US, 6346532[P]. 2002-02-12.
[6] 张 华,李 杨,陈仕杰,等. 米拉贝隆的合成方法: CN, 103896872[P]. 2014-07-02.
[7] 岑均达,黄 伟,李 强. 一种米拉贝隆及其中间体的制备方法: CN, 103387500[P]. 2013-11-13.
[8] 葛德培,吴其华. 米拉贝隆的合成方法: CN, 104230840[P]. 2014-12-24.
[9] KAWAZOE S, SAKAMOTO K, AWAMURA Y,etal. Alpha-form or beta-form crystal of acetanilide derivative: EP, 1440969[P]. 2004-07-28.
[10] THIRUMALAI R S, ESWARAIAH S, RAGHURAM S. Process for the preparation of 2-(2-aminothiazol-4-yl)-N-[4-(2-{[(2R)-2-hydroxy-2-phenylethyl]amino}ethyl)phenyl]acetamide monohydrochloride, its intermediates and polymorph thereof: WO, 2014132270[P]. 2014-09-04.
[11] LAL B, SHENOY G, DAVE R,etal. Novel process for preparation of mirabegron and its intermediate: WO, 2015162536[P]. 2015-10-29.
[12] 范文进,曾正英,吴之波,等. 米拉贝隆合成工艺改进[J]. 精细化工中间体, 2016,46(4):42-47.
[13] 毛龙飞,苑李双,颜茗彦,等. 米拉贝隆的合成研究[J]. 化学研究与应用, 2016,28(4):521-524.
[14] 章 磊,谭支敏,焦慧荣,等. 米拉贝隆有关物质的合成[J]. 中国医药工业杂志, 2014,45(1):9-12.
[15] 郑亚东,和 龙,贾志丹,等. 米拉贝隆和去羟基米拉贝隆的合成[J]. 山东化工, 2016,45(1):3-5,9.
(编辑 WJ)
Synthesis of Related Substances in Mirabegron
BAOYu-xian1,ZHANGWei2,CHENBo3,ZHANGQing-hua3,XUGuang-yu1*
(1. College of Chemistry and Chemical Engineering, Hunan Normal University, Changsha 410081, China; 2. School of Medicine, Hunan Normal University, Changsha 410013, China; 3. Hunan Fangsheng Pharmaceuticals Limited, Changsha 410205, China)
TQ463
A
1000-2537(2017)05-0051-08
2017-02-28
国家自然科学基金资助项目(20602010)
*通讯作者,E-mail:gyxu@hunnu.edu.cn
10.7612/j.issn.1000-2537.2017.05.008
猜你喜欢
食品科学技术学报(2022年6期)2022-12-15
化工与医药工程(2022年3期)2022-08-08
现代装饰(2020年8期)2020-08-24
作文小学中年级(2020年6期)2020-07-24
作文·小学低年级(2020年6期)2020-07-14
天然产物研究与开发(2018年8期)2018-09-10
中国资源综合利用(2016年7期)2016-02-03
郑州大学学报(工学版)(2015年1期)2015-03-24
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
火炸药学报(2014年5期)2014-03-20

- 湖南师范大学自然科学学报的其它文章
- 后终期阶段城市化水平演变特征及驱动力分析
——以广州市为例 - 湖南省植被覆盖遥感反演信息量化统计
- Fluorine-Substituted H4W10O32 as a Highly-Efficient Catalyst for Oxidative Degradation of Methyl Orange by Hydrogen Peroxide in Aqueous Solution Under Ultraviolet Light Irradiation
- 基于非参数分析方法的湖南省县域经济时空演变分析
- 生物炭的制备、改性及其环境效应研究进展
- 具有非线性发生率的多易感群体的传染病模型研究