
PDMS-PEG 核壳结构复合粒子的制备
2017-10-09 05:55孙迎迎刘喜军娄春华
合成树脂及塑料 2017年5期
孙迎迎,刘喜军,2*,娄春华,2
(1. 齐齐哈尔大学材料科学与工程学院,黑龙江省齐齐哈尔市 161006;2. 黑龙江教育厅复合改性材料重点实验室,黑龙江省齐齐哈尔市 161006)
PDMS-PEG 核壳结构复合粒子的制备
孙迎迎1,刘喜军1,2*,娄春华1,2
(1. 齐齐哈尔大学材料科学与工程学院,黑龙江省齐齐哈尔市 161006;2. 黑龙江教育厅复合改性材料重点实验室,黑龙江省齐齐哈尔市 161006)
采用溶液聚合法制备了以轻度交联的聚二甲基硅氧烷(PDMS)弹性粒子为核芯、端异氰酸酯基聚乙二醇(NCO-PEG)为壳层的PDMS-聚乙二醇(PEG)核壳结构复合粒子,并表征了其结构。结果表明:最佳反应时间为6 h;PDMS弹性粒子表面的硅羟基与NCO-PEG的端异氰酸酯基确实发生了化学反应,PEG被成功接枝到PDMS弹性粒子表面,PDMS-PEG中Si,O,C,N元素含量与理论值基本相符;PDMS-PEG为两相结构,具有明显的核壳结构,壳层厚度在130 nm左右;PDMS-PEG的初始热分解温度在300 ℃以上,可以满足聚乳酸增韧改性研究要求。
弹性粒子 聚二甲基硅氧烷 聚乙二醇 核壳结构
聚乳酸(PLA)具有优良的生物降解性、生物相容性、生物吸收性以及良好的力学性能和加工性能[1];但PLA的抗冲击性能较差,严重限制了其应用。为改善PLA的抗冲击性能,人们尝试各种方法对PLA的增韧改性进行研究和探索[2-4]。物理共混改性简单、经济且十分有效,被广泛应用于PLA的增韧改性[5],但PLA与大部分高分子材料不相容,简单的物理共混改性很难达到理想的增韧效果。软核硬壳型核壳结构复合粒子是近年发展起来的一种新型聚合物增韧剂,由于壳层聚合物的组成和结构可人为控制,因此,增强了增韧剂与PLA的相容性,极大改善了PLA共混物的综合性能[6]。目前,PLA增韧改性使用的增韧剂大多为不可生物降解、不相容材料,而采用可生物降解、相容材料增韧改性PLA还鲜有报道。
本工作以轻度交联的聚二甲基硅氧烷(PDMS)弹性粒子为核芯、端异氰酸酯基聚乙二醇(NCO-PEG)为壳层,采用溶液聚合法[7-8]制备了PDMS-聚乙二醇(PEG)核壳结构复合粒子,并对产物的组成、结构和形态进行了表征。
1 实验部分
1.1 主要试剂
PDMS按文献[9]制备。NCO-PEG按文献[10]制备。四氢呋喃(THF),分析纯,国药集团化学试剂有限公司生产。二月桂酸二丁基锡(DBTDL),二正丁胺,圴为分析纯;指示剂溴甲酚绿;丙酮,甲苯,乙醇,盐酸:圴为分析纯,市售。实验用水为三次蒸馏水。
1.2 PDMS-PEG核壳结构复合粒子的形成机理
采用乳液聚合法,以八甲基环四硅氧烷(D4)、四甲基四乙烯基环四硅氧烷(VD4)和甲基三乙氧基硅烷(MTES)为单体,在酸性条件下水解、缩聚合得到PDMS核乳液。PDMS核乳液破乳、干燥后分散到THF中,再加入NCO-PEG的THF溶液,通过异氰酸酯基(—NCO)与羟基(—OH)的加成反应,PEG在PDMS弹性粒子表面接枝形成壳层,得到PDMS-PEG核壳结构复合粒子,形成机理见图1。
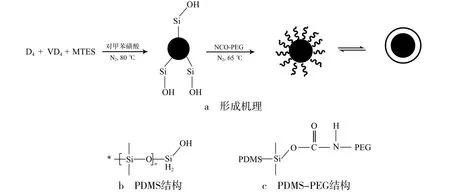
图1 PDMS-PEG核壳结构复合粒子结构及形成机理Fig.1 Structure and formation mechanism of PDMS-PEG core-shell composite particles
1.3 试样制备
将2 g PDMS和20 mL THF依次加入到配有搅拌桨、回流冷凝管和N2入口的250 mL三颈瓶中,待PDMS充分溶胀后,开动搅拌使之分散圴匀,N2保护,升温至65 ℃,加入0.1 mL DBTDL。将5 g NCOPEG和30 mL THF依次加入到100 mL圆底烧瓶中配成NCO-PEG的THF溶液,然后将其通过恒压滴定管在快速搅拌下缓慢滴加到三颈瓶中,滴加结束后继续反应6 h。将反应液移入旋转蒸发器,在70 ℃水浴中蒸出THF,得到固体产物。
以丙酮为萃取剂,采用索氏萃取器将固体产物萃取24 h,除去未接NCO-PEG,将提纯后的固体产物于50 ℃真空干燥12 h,即得到PDMS-PEG核壳结构复合粒子。
1.4 测试与表征
傅里叶变换红外光谱(FTIR)采用美国PE公司的Spectrum One型傅里叶变换红外光谱仪测试,KBr压片。X射线光电子能谱(XPS)采用美国Thermo Fisher公司的ESCALAB250Xi型X射线光电子能谱仪测试,Mg靶,Kα射线,真空度为1.33×10-6Pa。将PDMS-PEG核壳结构复合粒子溶于THF中,超声30 min使之分散圴匀,将溶液滴在覆有碳膜的铜网上,干燥后采用日本日立公司的H-7650型透射电子显微镜观察试样的形态。采用德国耐驰仪器制造有限公司的DSC204F1型差示扫描量热仪测试试样的热转变温度,N2气氛,升温速率为20℃/min,测试温度为-150~100 ℃。采用美国PE公司的Pyris Diamond SII型热重-差热综合热分析仪测试试样的耐热性能,N2气氛,升温速率为10 ℃/min,测试温度为室温至900 ℃。
2 结果与讨论
2.1 反应时间的确定
—OH和—NCO的加成反应是一个逐步进行的过程,可以通过检测反应体系中—NCO的含量来判断加成反应是否达到平衡,—NCO含量的变化可以直观反映加成反应的进程。采用甲苯-二正丁胺滴定法跟踪反应过程中—NCO含量随反应时间的变化。从图2可看出:反应初期,PDMS与NCO-PEG反应速率较快,特别在1~3 h时,—NCO含量快速下降,在4~6 h时,反应趋于平衡。这主要是由于反应初期,反应体系黏度较小,反应物浓度较高、扩散速率较快,加成反应速率较快,因此—NCO含量急剧下降。随着反应时间的延长、反应程度的提高,NCO-PEG的浓度降低,加之反应体系黏度增大,导致加成反应速率下降,—NCO含量变化趋缓。当反应时间超过6 h后,—NCO含量趋近于零,此时若继续延长反应时间,不但对提高加成反应进程意义不大,而且会导致一些副反应发生,因此,最佳反应时间为6 h。

图2 —NCO浓度随反应时间变化曲线Fig.2 —NCO group concentration as a function of reaction time
2.2 FTIR分析
从图3可以看出:PDMS在1 020,1 098 cm-1处存在环四硅氧烷开环后O—Si—O的伸缩振动吸收峰,3 443 cm-1处存在Si—OH的特征吸收峰;NCO-PEG在840 cm-1处存在亚甲基C—H的特征吸收峰,2 243 cm-1处存在—NCO的特征吸收峰;PDMS-PEG在843,1 018,1 107 cm-1处圴存在吸收峰,但Si—OH特征吸收峰完全消失,—NCO特征吸收峰明显减弱,说明PDMS与NCO-PEG确实发生了加成反应,PDMS弹性粒子表面的Si—OH与NCO-PEG分子链末端的—NCO发生了加成反应,生成了PDMS-PEG核壳结构复合粒子,即PEG被成功接枝到PDMS弹性粒子表面。

图3 PDMS-PEG的FTIRFig.3 FTIR spectra of PDMS-PEG
2.3 XPS分析
从图4可以看出:284 eV处为C1s光电子峰,530 eV处为O1s光电子峰,101 eV处为Si2p光电子峰,由于试样中N元素含量较少,因此,其光电子峰表现不明显。上述实验结果证明:PDMS中含有C,O,Si元素;NCO-PEG中含有C,O,N元素;PDMS-PEG中含有C,O,Si,N元素,说明PDMS与NCO-PEG的加成反应成功进行。

图4 PDMS-PEG的XPSFig.4 XPS spectra of PDMS-PEG
若PDMS与NCO-PEG加成反应进行完全,根据投料量和反应物的化学组成可以计算PDMSPEG核壳结构复合粒子中C,O,Si的含量。通过理论计算得到,PDMS乳胶粒子中,w(C),w(O),w(Si)分别为54.40%,24.20%,21.40%;PDMSPEG核壳结构复合粒子中,w(C),w(O),w(Si)分别为63.00%,31.00%,6.00%。通过对比发现,PDMS-PEG核壳结构复合粒子中C和O含量明显增加,并且C含量增加幅度较O含量稍大,相反Si含量明显减小,这是因为PEG中不含Si原子,PEG中C与O原子数之比为2∶1所致。
从表1可以看出:实验测得PDMS-PEG核壳结构复合粒子中w(C),w(O),w(Si)分别为61.82%,26.90%,10.50%,其规律性与理论计算结果完全相符。Si含量的实测值大于理论值,而C和O含量的实测值圴小于理论值。这可能是由于NCO-PEG投料量过大,导致部分NCO-PEG没有参与反应,使产物中C,O,Si含量的实测值与理论值存在一定差距。

表1 PDMS-PEG的元素组成实测值Tab.1 Elemental composition measured value of PDMS-PEG %
2.4 形态结构分析
从图5可以看出:PDMS-PEG复合粒子呈球状,分散圴匀,分布范围较窄,平圴粒径在0.8 µm左右,具有清晰的核壳结构,壳层厚度在130 nm左右。颜色较深的核芯部分为轻度交联的PDMS,颜色较浅的壳层部分为PEG大分子堆积体,两者通过氨基甲酸酯键结合形成一个整体。由先前的工作可知,反应物NCO-PEG的数圴分子量为3.5×104,倘若分子链完全伸直,长度约为205 nm,对照实验结果可知,PEG大分子链并未完全伸展。另外,壳层对核芯呈完全包覆状态,并且壳层厚度圴匀,说明PEG在壳层呈单分子层结构,进一步证明了NCO-PEG的相对分子质量分布较窄[10]。
2.5 差示扫描量热法(DSC)分析
从图6看出:PDMS在-119.85 ℃发生玻璃化转变,NCO-PEG在61.85 ℃出现熔融峰,该峰为PEG的熔点,PDMS-PEG在-117.13 ℃为核芯PDMS的玻璃化转变温度,60.15 ℃为壳层PEG的熔点。虽然PDMS与PEG通过化学键结合为一个整体,但PDMS-PEG的玻璃化转变温度、熔融峰并没有相互干扰。这说明PDMS与PEG确实发生了加成反应,PEG被成功接枝到PDMS表面,产物PDMS-PEG为两相结构,进一步证明其为核壳结构复合粒子。

图5 PDMS-PEG的透射电子显微镜照片(×20 000)Fig.5 TEM photo of PDMS-PEG

图6 PDMS-PEG的DSC曲线Fig.6 DSC curves of PDMS-PEG
2.6 耐热性能
从图7可以看出:随着温度的升高,PDMSPEG核壳结构复合粒子出现两次热分解过程:第一阶段质量损失温度为325~430 ℃,分解速率最大时的温度在400 ℃左右,质量损失约44%,主要是壳层中氨基甲酸酯键、PEG嵌段的分解与气化造成的;第二阶段质量损失温度为450~600 ℃,分解速率最大时的温度在550 ℃左右,质量损失约51%,主要是核层PDMS中Si—O—Si断裂、生成环状硅氧烷造成的。聚硅氧烷在高温惰性气氛中主要发生热解聚,热解聚的机理是:分子链内O原子的未共用电子对与邻近Si原子的3d空轨道配位,在高温或催化剂等作用下,Si—O—Si断裂、重排,产生小分子环状硅氧烷,如三元环、四元环、六元环、八元环等小分子环硅氧烷[11]。实验最终残留5%左右的非挥发物质,原因是硅橡胶的烧蚀过程是热解成硅质炭的过程,硅橡胶受热软化后,侧链先于主链断裂,因而主链上的Si将以炭的形式保留下来,形成炭化层。实验结果说明,PDMS-PEG核壳结构复合粒子热分解温度在300 ℃以上,完全能够满足PLA增韧改性研究要求。
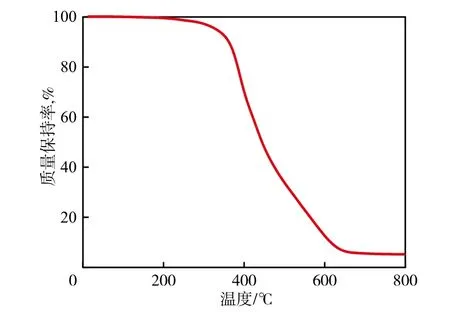
图7 PDMS-PEG的热重曲线Fig.7 TG curve of PDMS-PEG
3 结论
a)采用溶液聚合法合成的PDMS-PEG核壳结构复合粒子呈球状、分散性较好、分布范围较窄,其平圴粒径在0.8 µm左右,确实具有核壳结构。
b)PDMS与NCO-PEG的最佳反应时间为6 h。
c)PDMS与NCO-PEG的加成反应将NCOPEG成功接枝到PDMS弹性粒子表面。
d)PDMS-PEG核壳结构复合粒子的热分解温度在300 ℃以上,可以满足PLA增韧改性研究要求。
[1] 丁美春,刘漫,吴宁晶. ACR增韧聚乳酸的力学性能研究[J]. 塑料工业,2011,39(7):28-30.
[2] Song Xiaoli,Chen Ying,Xu Yuzhi,et al. Study on tough blends of polylactide and acrylic impact modifier[J]. Bioresources,2014,9(2):1939-1952.
[3] Rashmi B J,Prashantha K,Lacrampe M F,et al. Toughening of poly(lactic acid)without sacrificing stiffness and strength by meltblending with polyamide 11 and selective localization of halloysite nanotubes[J]. Express Polym Lett,2015,9(8):721-735.
[4] Qu Ping,Gao Yuan,Wu Guofeng,et al. Nanocomposites of poly(lactic acid)reinforced with cellulose nanofibrils[J].Bioresources,2010,5(3):1811-1823.
[5] Hassan A,Balakrishnan H,Akbari A.Polylactic acid based blends,composites and nanocomposites[J]. Adv Struct Mater,2013,18:361-396.
[6] 冯玉林,殷敬华,姜伟. 环氧基团功能化弹性体增韧聚乳酸的性能[J]. 高等学校化学报,2012,33(2):28-30.
[7] Zhao Xiaguo,Liu Wenfang,Li Ying,et al.Grafting of poly(ethylene glycol)onto nanometer silica surface by a one-step procedure[J]. Macromol Sci,2005,42(2):221-230.
[8] Chen Xiaohui,Pei Xu,Pan Xue,et al. Synthesis and characterization of silicone modified polyurethane surfactant useable at high temperature[J]. Macromol Sci,2013,50(2):703-708.
[9] 孙迎迎,刘喜军,娄春华. PDMS乳胶粒子的制备及其再分散性研究[J]. 齐齐哈尔大学学报,2015,36(6):65-69.
[10] 孙迎迎,刘喜军,娄春华. 端异氰酸酯基聚氨酯预聚体的合成及其应用研究[J]. 广州化工,2016,44(3):49-53.
[11] 陈循军,崔英德. B(C6F5)3对聚二甲基硅氧烷热降解行为的影响[J]. 化工新型材料,2010,38(4):71-73.
Preparation of PDMS-PEG core-shell composite particles
Sun Yingying1, Liu Xijun1,2, Lou Chunhua1,2
(1. College of Materials Science and Engineering, Qiqihar University, Qiqihar 161006, China;
2. Key Laboratory of Composition and Modi fi cation Materials, College of Heilongjiang Province, Qiqihar 161006, China)
Polydimethyl silocane(PDMS)-polyethylene glycol(PEG)core-shell composite particles were prepared via solution polymerization,which consist of lightly crosslinked PDMS elastic particles as core and isocyanate-terminated PEG(NCO-PEG)as shell. The structure of the particles were characterized. The results show that the optimal reaction time is 6 h. Silicon hydroxyl group on the surface of PDMS elastic particles reacts with the terminal isocyanate group of NCO-PEG. PEG is successfully grafted onto the surface of PDMS. The contents of Si,O,C and N in PDMS-PEG are equal to the theoretical values. There is obvious two-phase coreshell structure in PDMS-PEG and the average shell thickness is 130 nm. The initial thermal decomposition temperature of PDMS-PEG is above 300 ℃,which meets the requirements of polylactic acid toughening modification.
elastic particle; polydimethyl silocane; polyethylene glycol; core-shell structure
TQ 050.4+3
B
1002-1396(2017)05-0041-05
2017-04-27;
2017-07-06。
孙迎迎,女,1988年生,硕士研究生,主要研究方向为高分子材料制备与应用。联系电话:18238770430;E-mail:1078556373@qq.com。
黑龙江省自然科学基金资助项目(EZ01343)。
*通信联系人。E-mail:liuxijun2002@163.com。
猜你喜欢
发光学报(2021年7期)2021-07-23
组织工程与重建外科杂志(2018年6期)2018-01-12
西安工程大学学报(2016年6期)2017-01-15
发光学报(2016年10期)2016-11-19
中国塑料(2016年1期)2016-05-17
中国塑料(2015年3期)2015-11-27
中国塑料(2015年2期)2015-10-14
中国塑料(2015年10期)2015-10-14
中国塑料(2014年12期)2014-10-17
中国塑料(2014年12期)2014-10-17
