
应用高通量测序技术鉴别中国输入性黄热病毒野毒株与疫苗株
2017-09-28 05:35:37刘林张益李阿茜张硕张全福李川严延生马学军梁米芳李德新
中华实验和临床病毒学杂志 2017年4期
刘林 张益 李阿茜 张硕 张全福 李川 严延生 马学军 梁米芳 李德新
102206 北京,中国疾病预防控制中心病毒病预防控制所(刘林、张益、李阿茜、张硕、张全福、李川、马学军、梁米芳、李德新);350001 福州,福建省疾病预防控制中心(严延生)
·技术方法·
应用高通量测序技术鉴别中国输入性黄热病毒野毒株与疫苗株
刘林 张益 李阿茜 张硕 张全福 李川 严延生 马学军 梁米芳 李德新
102206 北京,中国疾病预防控制中心病毒病预防控制所(刘林、张益、李阿茜、张硕、张全福、李川、马学军、梁米芳、李德新);350001 福州,福建省疾病预防控制中心(严延生)
目的鉴别2016年3月我国3例输入性黄热病例为2016年安哥拉流行的黄热病毒野毒株感染,还是黄热病毒疫苗相关疾病,亦或是二者的混合感染。方法应用IonTorrent PGM测序平台,对3例黄热病病例血液或尿液标本进行高通量测序,对所获得的3株黄热病毒序列进行基因组特征分析。分析黄热病毒野毒株与疫苗株核酸序列差异,以同源性较低区域序列作为参考序列将高通量测序获得的短序列做回贴和比对。结果从3份黄热患者标本中获得了部分黄热病毒基因组,其中从第二例患者标本中获得了全长蛋白编码区序列。3株病毒与2016年从中国第一例输入性黄热病例中分离的病毒株CNYF01R/2016株和来自安哥拉的1971年毒株Angola71株具有极高的相似性,都属于安哥拉型黄热病毒。分析得到5段黄热病毒野毒株与疫苗株核酸序列差异较大的区域,在这5段区域中,3份标本测序获得短序列与野毒株均有多个匹配,而与疫苗株则没有匹配。结论3例黄热患者的血液或尿液标本中未检测到黄热病毒疫苗株17D株基因组序列,高通量测序证实3人均为2016年安哥拉流行的黄热病毒野毒株感染。
黄热病是由黄热病毒引起的一种急性传染病,主要通过蚊媒传播。黄热病毒是黄病毒科黄病毒属的代表种,它起源于非洲,最早的分离株(Asibi株)分离于1927年加纳的黄热病患者血液接种的恒河猴[1]。此后,黄热病在奴隶贸易时期由非洲传入美洲[2],另外在欧洲也有过多次黄热病的流行。目前,虽已具有有效的减毒活疫苗(17D疫苗株)[3],但是在非洲、南美等很多区域黄热病依然构成严重的公共卫生威胁。2016年,非洲安哥拉暴发严重的黄热病疫情,并首次输入到我国,累计报道了11例输入性病例。这些病例均为中国公民,有的去安哥拉务工前未接种过黄热疫苗,安哥拉黄热病疫情暴发后,在当地采取了应急接种,接种后不久,出现发热等疑似黄热病症状回国接受治疗。有报道称黄热病减毒疫苗可造成严重疾病[4],这些黄热病患者是由于感染安哥拉流行的黄热病毒野毒株,还是由于所接种的黄热病减毒活疫苗造成的,是需要回答的的公共卫生问题。患者在本研究中,我们采用高通量测序技术,对3份患者样本进行了黄热病毒野毒株和疫苗株的鉴别。
1 材料与方法
1.1研究对象3例黄热病疑似病例的标本由福建省疾病预防控制中心采集,并送检至中国疾病预防控制中心。3例疑似病例均是在安哥拉的中国工作人员,此前都没有接种过黄热疫苗,安哥拉黄热病流行暴发后才在当地接种17D减毒活疫苗,而后在当地出现发热等疑似黄热病症状,回国治疗。3例病例的采样信息见表1。样品经冷链运送至中国疾控中心病毒病预防控制所后,立即进行相关实验。
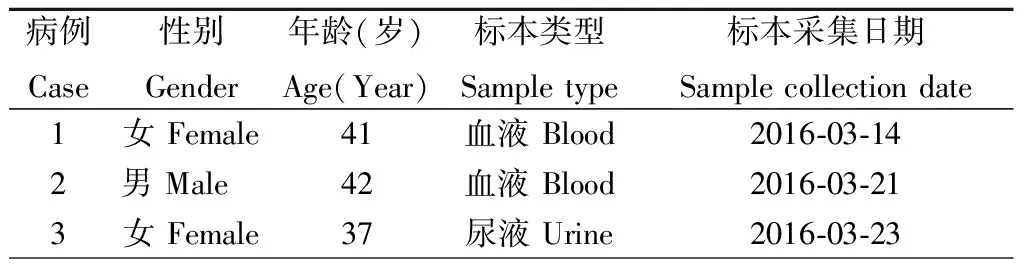
表1 病例信息
1.2RNA提取使用Qiagen 公司的RNeasy Plus Mini Kit提取患者标本总RNA。每份标本取100 μl严格按照试剂盒说明书进行操作。先将100 μl标本与350 μl Buffer RLT Plus混合后,移入去除人基因组的纯化柱,离心后取滤过液体,加入250 μl无水乙醇并混匀。将上一步的混合液移入核酸提取柱中并离心,再经过RW1和RPE洗涤,最后用40 μl无RNase水洗脱。提取后RNA立即进行后续实验或保存于-80 ℃冰箱备用。
1.3核酸扩增及定量利用Qiagen公司的REPLI-g WTA Single Cell Kit对所提取RNA进行全转录组的等温扩增。先经过去除人基因组、逆转录、连接等步骤,最后利用随机引物进行多重置换扩增(MDA)得到扩增后的核酸。然后使用Thermo Fisher公司的Qubit® 3.0荧光定量仪对扩增后的核酸进行定量。
1.4高通量测序取100 ng上一步扩增后的核酸,使用Thermo Fisher公司的Ion XpressTMPlus Fragment Library Kit按试剂盒说明书进行测序文库构建(200 bp)。然后使用Ion Library Quantitation Kit对构建好的测序文库进行qPCR定量。然后使用Ion PGMTMHi-QTMOT2 Kit,在Ion OneTouchTM2 System上进行油包水反应,制备测序模板。最后使用Ion PGMTMHi-QTMSequencingKit和Ion 318TM芯片,在Ion PGMTM测序仪上进行测序。
1.5测序数据分析使用CLC Genomics Workbench 8.5.1软件分析高通量测序结果。分别以黄热病毒17D疫苗株(GenBank号X03700.1)和2016年安哥拉黄热病流行株CNYF01R/2016株(GenBank号KU921608.1)作为参考序列,将3份标本的高通量测序得到的短序列往两个参考序列上分别做回贴(mapping)。将得到的黄热病毒序列在NCBI数据库中做BLAST分析,然后使用MEGA6.06软件对新获得黄热病毒序列与GenBank中黄热病毒各基因型的典型毒株序列进行Muscle法多序列比对,然后使用邻接法进行系统发育分析,采用Yokose病毒作为外群,Bootstrap检验1 000次验证进化树有效性。
1.6疫苗株与野毒株的鉴别使用Simplot 3.5.1软件对本次安哥拉黄热病流行的野毒株CNYF01/2 016株与黄热病毒17D疫苗株进行基因组各区段的相似性对比,窗口长度200 bp,步长20 bp。根据上一步两序列相似性比对结果,找出5段相似度较低的序列区段。将这5段相似度低的序列作为参考序列,使用CLC Genomics Workbench 8.5.1软件,设置不同的匹配参数,通过匹配的结果来分析样本中的黄热病毒是疫苗株还是野毒株,或者是两者的混合感染。
2 结果
2.1高通量测序应用IonTorrent PGM测序平台,对3份黄热疑似病例标本进行高通量测序和初步序列分析。从病例2的标本中几乎测得了该株黄热病毒全长序列,病毒基因组覆盖度达99.25%,其中包括了完整的开放读码框区域。从病例1和病例3的标本中仅测得了黄热病毒的部分基因组序列,基因组覆盖度分别为26.68%和59.89%(表2)。
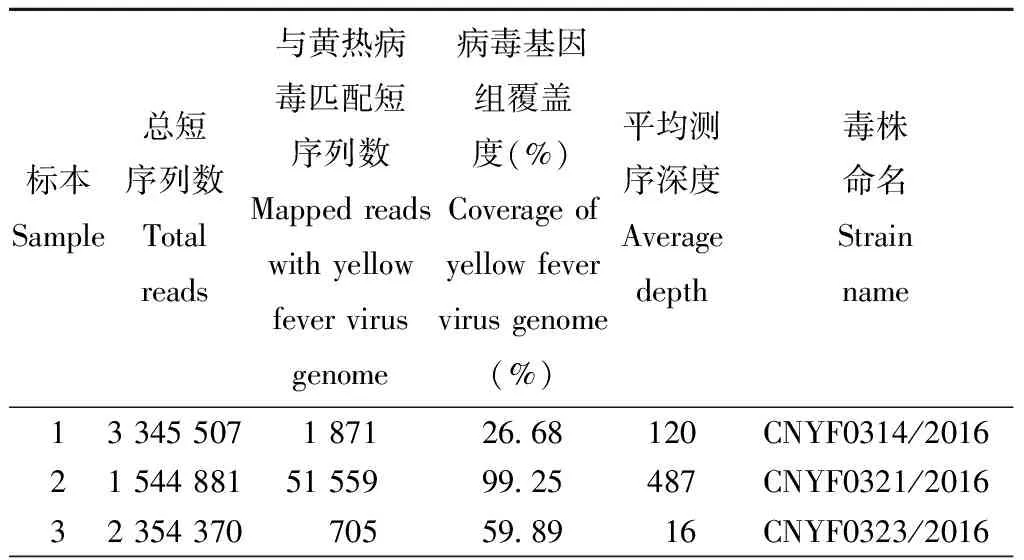
表2 3份标本高通量测序数据
2.23株病毒的基因特征将从3份标本中获得的毒株序列上传至GenBank,编号分别为KY873606、KY873607、KY873608。根据3株黄热病毒均有覆盖的PreM蛋白区域(397 ~577 nt),结合黄热病毒其他基因型的典型毒株,进行系统发育分析。与新获得的3株黄热病毒相似度最高的毒株是2016年从中国第一例输入性黄热病例中分离得到的CNYF01R/2016株,它们都属于安哥拉型黄热病毒,起源于安哥拉1971年的黄热病流行(图1)。
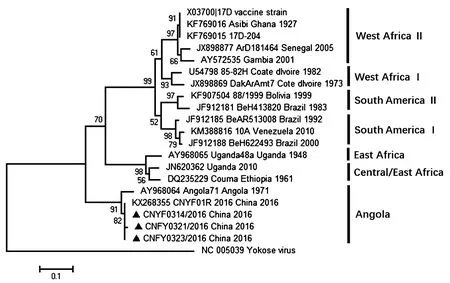
注:▲新获得的3株黄热病毒图1 部分PreM基因(397~577 nt)的系统进化分析Note: ▲Three newly obtained yellow fever virus strainsFig.1 Phylogenetic analysis of partial PreM gene (397~577 nt)
2.3疫苗株与野毒株的基因组的差异比较分析使用Simplot软件将中国2016年的黄热病毒流行株CNYF01R/2016株(KX268355)作为参考序列,比较其与黄热病毒野毒株Angola71株(AY968064)和疫苗株17D株(X03700)的基因组各区段相似性。从图2中可以看出,中国2016年的黄热病毒流行株与安哥拉1971年的流行株Angola71株在基因组的各区段相似性都非常高,基本都在95%以上,而它们与疫苗株17D株差异较大。表3中列出了5段疫苗株与野毒株的基因组核酸序列差异较大的区段。
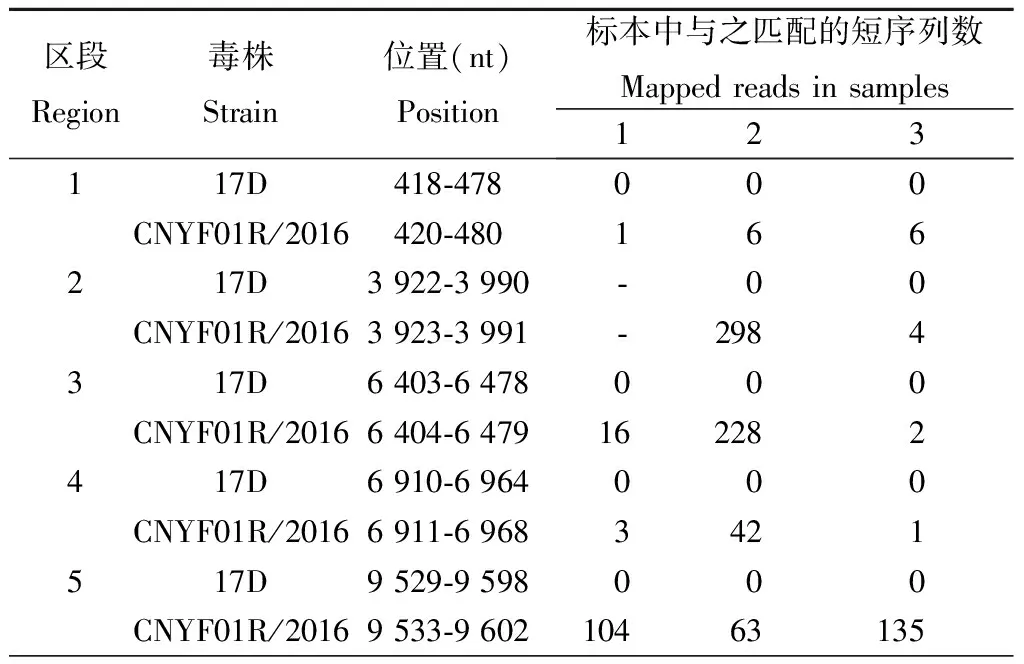
表3 疫苗株与野毒株的基因组核酸序列差异较大区段
2.4疫苗株与野毒株鉴别以表3中5个区段的两株病毒序列分别作为参考序列,使用CLC软件将3个标本的测序数据分别与之比对,得到匹配的短序列数见表2。除病例1标本中没有病毒序列覆盖区段2,在其余的区段3个标本都有短序列覆盖。从表3中可以看出,在5个差异较大区段,3例标本中均未出现与疫苗株17D株相匹配的短序列,而都有与野毒株相匹配的序列。另外,我们还特别注意到在区段4中,CNYF01R/2016株的6 944 nt~6 946 nt的位置对比17D疫苗株插入了3个碱基CAC,导致在蛋白序列中插入一个组氨酸(H)。而在3份标本中,与该区段相匹配的短序列都存在这3个碱基的插入。由此可以推断,本研究检测的3份标本中,没有发现疫苗株17D株病毒核酸,此3例黄热病例均为2016年流行的黄热病野毒株感染。
3 讨论
本研究应用高通量测序技术,对2016年3月份3例黄热病输入性病例进行了病毒基因组解析,并应用相关生物信息学分析软件对测序获得的黄热病毒序列是属于疫苗株还是野毒株,亦或两者混合感染进行了精准鉴别。在这些患者的血液或尿液中检测到2016年流行的黄热病毒野毒株核酸,没有检测到疫苗株病毒核酸。因此,可认为导致患者发病的是黄热病野毒株而不是疫苗株,可能是患者应急接种疫苗后,尚未产生足够的保护力的情况下,即被黄热病毒野毒株感染而发病。
本研究采用的高通量测序技术比传统的一代测序技术在很多方面有着很大的优势,可以获得样本中数百万甚至上千万的短序列,可以查明每个短序列的来源,不仅可以检测出样本中的主要病毒株,还可判定检测样本中的混合感染情况[5]。在检测证实这几位黄热患者感染是黄热病毒野毒株的同时,明确了他们的样本中没有黄热病疫苗株病毒核酸。
本研究在鉴别混合感染情况时,为防止来自不同毒株的序列拼接在一起,未对短序列进行contig拼接,而是对每个短序列单独进行分析。然而对每个短序列进行BLAST分析需要非常强大的计算机运算能力和非常长的运算时间,更重要的是,在两个毒株相似性高的区域出现的短序列,在分析时可能出现排在第一位的相似序列与其他相似序列区分度不明显的现象,容易得出错误的结论。因此本研究将高通量测序获得的短序列采取贴到两毒株差异较大的区域,并结合特异性突变位点的分析,达到了很好的鉴别效果。该研究方法也可以应用到利用高通量测序鉴别其他病毒的混合感染。
17D减毒株制备的黄热病疫苗可以有效预防黄热病。虽然有过黄热病减毒疫苗引起YEL-AND、YEL-AVD等严重疾病的报道[4],但是总体上出现黄热病疫苗相关疾病的几率非常低,在全球范围,每接种100万次黄热疫苗,会出现从0.09次(巴西)到2.5次(美国)黄热疫苗相关疾病[6]。接种黄热病疫苗10 d后,90%以上接种者可以获得免疫力;接种黄热疫苗30 d后,99%以上接种者可以获得免疫力[7]。本研究的3位黄热病患者在前往安哥拉之前未接种黄热病疫苗,虽然当地黄热病疫情暴发之后应急接种了疫苗,但是感染很可能发生在接种后尚未产生足够免疫力或患者接种疫苗时已经感染,处于潜伏期(3~6 d,最多14 d)未发病。因此前往黄热病流行的国家或地区旅行或者工作的人,应严格按照检疫条例要求,在到疫情地之前至少10 d接种疫苗,到疫区后应该提高防蚊意识并采取相应措施。
[1] Stokes A, Bauer JH, Hudson NP. The transmission of yellow fever to Macacus rhesus. 1928[J]. Rev Med Virol, 2001, 11(3): 141-148.
[2] Barrett AD, Monath TP. Epidemiology and ecology of yellow fever virus[J]. Adv Virus Res, 2003, 61: 291-315.
[3] Theiler M, Smith HH. The effect of prolonged cultivation in vitro upon the pathogenicity of yellow fever virus[J]. J Exp Med, 1937, 65(6): 767-786.
[4] Lang J, Zuckerman J, Clarke P, et al. Comparison of the immunogenicity and safety of two 17D yellow fever vaccines[J]. Am J Trop Med Hyg, 1999, 60(6): 1045-1050.
[5] 张静娜,王译葵,蒋岩,等. 高通量测序技术用于丙型肝炎病毒感染溯源调查[J]. 中华预防医学杂志, 2016,50(6):530-534. doi.org/10.3760/cma.j.issn.0253-9624.2016.06.011
[6] Rosenthal S, Chen R. The reporting sensitivities of two passive surveillance systems for vaccine adverse events[J]. Am J Public Health, 1995, 85(12): 1706-1709.
[7] 国家卫生和计划生育委员会国家质检总局. 黄热病防控方案(2016 年版)[J]. 国际流行病学传染病学杂志, 2016, 6,43(3): 150-152. doi:10.3760/cma.j.issn.1673-4149.2016.03.002
DistinguishvaccinestrainandwildtypestrainofyellowfevervirusimportedtoChinausinghigh-throughputsequencingtechnology
LiuLin,ZhangYi,LiAqian,ZhangShuo,ZhangQuanfu,LiChuan,MaXuejun,LiangMifang,LiDexin
NationalInstituteforViralDiseaseControlandPrevention,ChineseCenterforDiseaseControlandPrevention,Beijing102206,China(LiuL,ZhangY,LiAQ,ZhangS,ZhangQF,LiC,MaXJ,LiangMF,LiDX);FujianProvincialCenterforDiseaseControlandPrevention,Fuzhou350001,China(YanYS)
LiDexin,Email:lidx@chinacdc.cn
ObjectiveTo identify whether the three imported yellow fever cases in China in March 2016 were infections by wild type strain of yellow fever virus in Angola in 2016, vaccine-associated disease or co-infection of both.MethodsSequences of three yellow fever virus strains were obtained by high-throughput sequencing with IonTorrent PGM platform from blood or urine samples of three yellow fever cases, and their genomic characteristics were analyzed. Then the regions with relatively great difference between the wild type strain and 17D vaccine strain were identified, and then served as the reference sequences when mapping the reads obtained by high-throughput sequencing.ResultsPartial yellow fever virus genomes were obtained from three samples of yellow fever patients, among them a full length coding region sequence was gained in sample 2. Comparing the genome sequences, the three newly obtained strains of yellow fever virus were highly similar to strain CNYF01R / 2016 which was isolated from the first imported yellow fever case to China in 2016 and strain Angola 71 from Angola in 1971, and they all belonged to Angola genotype of yellow fever virus. In this study, we found five regions in yellow fever virus genomes with great diversity between the vaccine strain and the wild type strain. In these five regions, a number of short reads obtained by high-throughput sequencing of the three samples were mapped to the sequence of wild type virus, while no short reads matched the vaccine strain.ConclusionsThere were no viral nucleic acid of 17D vaccine strain in the blood or urine samples of these three cases of yellow fever. They are all infected by wild type strains of Angola in 2016.
Yellow fever virus; High-throughput sequencing
黄热病毒;高通量测序
2017-04-21)
(本文编辑:唐浏英)
李德新, Email:lidx@chinacdc.cn
10.3760/cma.j.issn.1003-9279.2017.04.017
科技部改革发展专项,寨卡疫情防控科技攻关应急专项 [国资发2016(273)号)
Fundprograms: ZIKA Special Project of the MOST Reform and Development Project [273(2016)]
猜你喜欢
国际太空(2023年1期)2023-02-27 09:03:42
透析与人工器官(2020年1期)2020-11-16 01:42:34
铁道通信信号(2019年8期)2019-10-10 05:06:00
中国外汇(2019年10期)2019-08-27 01:58:14
世界知识(2017年10期)2017-06-03 10:57:15
中国发展观察(2017年8期)2017-04-26 03:51:50
家庭用药(2016年5期)2016-05-14 16:49:30
健康管理(2016年5期)2016-05-14 09:19:37
甘肃医药(2016年3期)2016-03-09 19:10:59
城市轨道交通研究(2015年3期)2015-02-27 11:01:37
