
Ⅲ型胶原纤维肾小球病的临床及病理特征分析
2017-09-22 08:37:09龚劭敏骆伟丽刘学光丁小强
中国临床医学 2017年4期
龚劭敏,骆伟丽,金 是,刘学光,丁小强,刘 红*
1. 复旦大学附属中山医院肾内科,上海市肾病与透析研究所,上海市肾脏疾病与血液净化重点实验室,上海 2000322. 复旦大学基础医学院病理学系,上海 200032
·短篇论著·
Ⅲ型胶原纤维肾小球病的临床及病理特征分析
龚劭敏1,骆伟丽1,金 是1,刘学光2,丁小强1,刘 红1*
1. 复旦大学附属中山医院肾内科,上海市肾病与透析研究所,上海市肾脏疾病与血液净化重点实验室,上海 2000322. 复旦大学基础医学院病理学系,上海 200032
目的: 分析Ⅲ型胶原纤维肾小球病患者的临床和病理资料,提升该病诊治水平。方法: 回顾性分析2007年3月至2015年6月肾活检患者的病例资料,进一步分析其中Ⅲ型胶原纤维肾小球病患者的临床和病理资料。结果: 共4例Ⅲ型胶原纤维肾小球病患者,其中2例男性,2例女性;年龄(45.5±10.7)岁。4例患者均无家族史,均无肾外累及。就诊时最常见临床表现为蛋白尿、高血压、肾功能不全。肾活检病理改变多样,可表现为膜增生样改变、结节性病变、局灶性节段性肾小球硬化;其中1例同时伴有IgA肾病。1例患者肾功能轻度减退伴少量蛋白尿,肾功能及尿蛋白定量长期稳定;2例患者糖皮质激素联合免疫制剂治疗有效,肾功能较前好转,蛋白尿部分缓解;1例患者进展至终末期肾病,接受血液透析治疗。结论: Ⅲ型胶原纤维肾小球病呈散发性,可表现为蛋白尿、高血压、肾功能减退;肾活检光镜病理表现多样,容易漏诊;免疫组织化学和电镜检查有助于确诊。
Ⅲ型胶原纤维肾小球病; 肾组织病理; 治疗
Ⅲ型胶原肾小球病亦称为胶原纤维肾小球病(collagen fibrotic glomerulalopathy,CG),是少见的特发性肾小球疾病,为常染色隐性遗传性疾病。正常肾小球基底膜和系膜区胶原为Ⅳ型,Ⅲ型胶原分布于肾间质和间质血管上,肾小球中无Ⅲ型胶原纤维。1979年,日本学者首先报道1例常染色体隐性遗传性疾病——甲髌综合征(nail-patella syndrome,NPS)病例,患者肾小球中可见胶原纤维异常沉积[1],此前后均有该现象报道[2-3]。1988年,Taguchi等[4]证实沉积的胶原纤维为Ⅲ型。1995年,世界卫生组织在肾小球疾病分类中列入了CG。
本研究通过回顾性分析本院确诊的4例散发Ⅲ型胶原肾小球病患者病例资料,并复习文献,以期帮助临床更好地了解这种少见的肾小球疾病。
1 资料与方法
1.1 入院资料 病例1:男性,30岁,蛋白尿10年,血肌酐(SCr)升高10 d,于2011年4月入院。患者10年前无明显诱因出现眼睑、双下肢水肿,血压升高,尿蛋白定量3.0 g/24 h。予泼尼松60 mg/d、雷公藤多苷10 mg(每日3次)及卡托普利25 mg/d、双嘧达莫50 mg/次(每日3次)治疗3个月后尿蛋白(-);泼尼松和雷公藤多苷减量,1年后停药,尿蛋白0.1~0.15 g/24 h。4年前查尿蛋白1.0 g/24 h,予泼尼松30 mg/d, 雷公藤多苷20 mg(每日3次)治疗, 2个月后尿蛋白(-),1年后停药。2008年,患者体检发现SCr 120 μmol/L,予泼尼松60 mg/d, 雷公藤多苷20 mg(每日3次)治疗,6个月后自行停药。此次因腹泻后查尿蛋白(),SCr 175 μmol/L入院;入院体格检查示,血压140/90 mmHg(1 mmHg=0.133 kPa),无其他阳性体征。入院后临床资料见表1。
病例2:女性,53岁,眼睑、双下肢水肿2年,蛋白尿、血尿4个月,于2010年5月入院。患者2年前出现晨起眼睑水肿,并逐渐出现下肢水肿伴腰酸,自行缓解,未就诊。入院前4个月水肿加重,尿蛋白(+),红细胞(),白细胞()。未出现其他肾外症状,血压正常,无其他疾病史,无阳性体征。入院后临床资料见表1。
病例3:女性,52岁,腰酸伴颜面水肿1年,SCr 升高4 d,于2015年3月入院。患者腰部酸痛伴晨起颜面部水肿1年,未就诊。入院前4 d于门诊查尿蛋白(),红细胞(±);尿素氮(BUN)18.0 mmol/L,SCr 243 μmol/L,尿酸(UA) 480 μmol/L;白蛋白(Alb) 32 g/L,血红蛋白(Hb)102 g/L。入院体格检查示,血压 156 /72 mmHg,余无阳性体征。入院后临床资料见表1。
病例4:男性,47岁,蛋白尿伴血压、SCr升高3年,于2015年6月入院。患者3年前查尿蛋白(),SCr 152 μmol/L,血压140/100 mmHg,口服氨氯地平、缬沙坦;随后SCr进行性升高伴下肢水肿。2个月前查SCr 362 μmol/L,因经济原因保守治疗。2周前因水肿加重入院。体格检查示,血压145/95 mmHg,颜面部、双下肢凹陷性水肿,无其他阳性体征。入院后临床资料见表1。
1.2 肾组织病理 病例1:光镜下,11个肾小球中有2个球性硬化,余肾小球体积增大、呈分叶状,肾固有细胞100~120个/球,内皮增生,系膜细胞中重度增生,系膜基质中重度增多,系膜区可见嗜复红物沉积,外周袢见双轨征,球内可见炎细胞浸润>5个/球,部分袢与球囊壁粘连,可见囊壁纤维化。小管间质病变轻,细动脉玻璃样变。免疫荧光显示,C3在系膜、血管袢弥漫分布。光镜诊断膜增生性肾炎(图1A)。电镜示,电子致密物在系膜区中等量沉积,在内皮下区、部分上皮下区少量沉积;系膜细胞节段性增生伴基质明显增多,部分节段向内皮下插入;系膜区、内皮下区均见较多胶原丝样纤维状物质沉积,致基膜明显增厚、管腔狭窄;足突广泛消失伴中度微绒毛形成。
病例2:光镜示,7个肾小球中有3个球性硬化,余小球系膜细胞轻度增生,系膜基质轻度增多。免疫荧光阴性。光镜诊断局灶节段增生伴硬化性肾炎(图1B)。电镜见部分血管袢内皮下区大量纤维束沉积、平行排列,伴系膜区少量沉积,系膜细胞节段性增生伴基质增多(图2A、2B),上皮细胞足突较广泛融合伴少量微绒毛形成。
病例3:光镜示,8个肾小球中有4个球性硬化,1个节段硬化,1个可见小细胞性新月体;系膜基质中度增多,系膜细胞中度增生,40%~50%小管萎缩,间质纤维化(),中等量炎细胞浸润聚集成团,血管壁增厚。免疫荧光示,IgA呈颗粒状、团块状在系膜区分布。光镜诊断为IgA肾病(图1C)。电镜示,系膜区轻度基质增多,小球中毛细血管袢基膜轻度增厚,基膜内可见大量平行排列的纤维状结构,可见横纹(图2C),足细胞足突广泛消失。
病例4:光镜示,17个肾小球中有10个球性硬化,余小球系膜基质重度增多,大量结节形成。大于75%的小管萎缩,间质纤维化(),大量炎细胞浸润聚集成团,血管壁明显增厚。刚果红染色(-),免疫荧光阴性。光镜示结节样肾小球硬化(图1D)。电镜示,系膜区基质大量增多,内有大量纤维状结构沉积,多呈纵行排列;高倍镜下有明暗间隔条纹,基膜结构不清,上皮足突广泛消失。

表1 4例患者入院后临床资料
*估算肾小球滤过率(estimated glomerular filtration rate, eGFR):使用MDRD公式计算. C3 参考范围0.79~1.52 g/L,C4 参考范围0.16~0.38 g/L

图1 Ⅲ型胶原肾小球病病理检查光镜结果
A: 膜增生性肾炎(H-E染色);B:局灶节段增生伴硬化性肾炎(Masson染色);C: IgA肾病(H-E染色);D: 结节样肾小球硬化(H-E染色). Original magnification: ×400
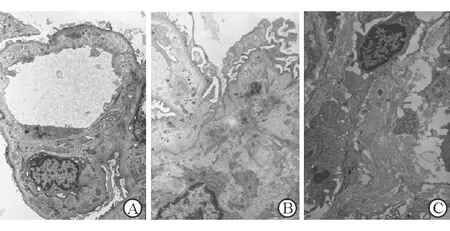
图2 Ⅲ型胶原肾小球病病理检查电镜结果
A:纤维束血管袢内皮下区平行排列; B:纤维束在系膜区少量沉积; C:基膜内大量纤维状结构平行排列. Original magnification: ×3 000(A),×5 000(B),×9 000(C)
2 结 果
病例1:泼尼松60 mg/d治疗1个月后复查尿红细胞(+),尿蛋白0.53 g/24 h, BUN 10.2 mmol/L,SCr 169 μmol/L,UA 567 μmol/L。泼尼松逐步减量,2011年5月至2012年5月随访过程中,尿蛋白0.3~0.4 g/24 h,但SCr逐步升高。治疗12个月后,泼尼松减量至10 mg/d,尿蛋白0.29 g/24 h、SCr 222 μmol/L,加用雷公藤多苷片20 mg (每日2次)治疗;1个月后,SCr降至152 μmol/L;随访中肾功能稳定,但尿蛋白逐渐增加至3.4 g/24 h,加用霉酚酸酯治疗;3个月后,蛋白尿降至1.2 g/24 h。继续随访2年,SCr稳定于150 μmol/L。
病例2:贝那普利10 mg/d治疗。随访7年,患者肾功能正常,尿蛋白<0.3 g/24 h。
病例3:患者HBsAg、HBeAb、HBcAg阳性,HBV-DNA阴性,予恩替卡韦抗病毒治疗2周后,予甲泼尼龙48 mg(每日1次),同时环磷酰胺0.6 g静脉滴注;1个月后随访,尿蛋白定量3.58 g/24 h,SCr 225 μmol/L。患者使用免疫抑制剂后反复感染,甲泼尼龙逐步减量至停药。随访1年,SCr波动于230 μmol/L左右、尿蛋白定量约3 g/24 h。
病例4:予控制血压、利尿消肿等到对症治疗后,患者水肿无明显好转;肾穿刺活检1个月后随访,SCr 654 μmol/L,行动静脉内瘘成形术;2个月后开始行血液透析治疗。
3 讨 论
Ⅲ型胶原肾小球病是Ⅲ型胶原纤维在肾小球中异常沉积引起的肾小球疾病,为一种罕见病。目前大部分该病的病例报道来自亚洲,主要来自日本学者,我国有少量病例报告[5],欧洲、南美洲均有个案报告[6-11]。但该病是否与地域或人种相关,目前仍不明确。由于Ⅲ型胶原纤维肾小球病最初在研究NPS患者时发现,且有在一个家系中有多个Ⅲ型胶原纤维肾小球病患儿的情况,故认为该病为常染色隐性遗传性疾病[12-13]。但在成人患者中,大部分为散发病例,无家族史,且患者发病年龄、性别差异无统计学意义。
肾脏内Ⅲ型胶原的来源目前有两种假说:一种为是内源性,通过对α平滑肌肌动蛋白的免疫组织化学染色发现,患者系膜区系膜细胞被激活,系膜细胞可能是肾内Ⅲ型胶原的主要来源[14];另一种假说与机体系统性Ⅲ型胶原合成和降解失衡有关,在部分患者中观察到血清Ⅲ型前胶原水平和血清透明质酸水平明显升高,由此推断过多的Ⅲ型胶原前体进入血循环并沉积在肾小球而致病[13,15-16]。本中心报道的2例男性、2例女性成人患者年龄30~53岁,均无家族史,无系统性疾病。因此认为,在成人患者中,遗传性因素可能并非主要的发病机制。
水肿、蛋白尿是Ⅲ型胶原肾小球病最常见的临床表现,部分患者表现为肾病综合征,可伴镜下血尿。肾功能减退及高血压也很常见[7]。肾脏病理改变多样,多数患者肾组织病理表现为肾小球体积增大,系膜区增宽,系膜基质增多和毛细血管袢增厚,部分可呈分叶状伴“双轨形成”,甚至表现为膜增生样改变;基底膜内,系膜区大量碘酸雪夫氏(PAS)染色弱阳性物质沉积[6,17-18],刚果红染色阴性;一般免疫荧光染色示IgG、IgA、C1q、C3阴性,IgM可呈非特异性沉积[5,19]。但是,Fukami等[20]报道了1例免疫荧光结果呈“满堂亮”的胶原纤维Ⅲ肾小球病的病例。本研究报道的4例患者病理表现各不相同:1例为C3肾病同时伴有胶原Ⅲ沉积,1例为IgA肾病伴胶原Ⅲ沉积,还有1例为结节样肾病;3例病理表现均罕见。国内目前仅陈惠萍等[5]报道1例IgA肾病病例,免疫荧光见IgA在系膜区沉积,后电镜证实合并IgA肾病。目前国内尚无C3肾病同时伴有胶原Ⅲ沉积肾小球疾病的报道。Ⅲ型胶原肾小球病必须依靠电镜检查和免疫组织化学染色进行确诊:电镜下可见基底膜内疏松层和系膜区大量弥漫沉积的胶原,呈束装分布,纤维直径40~100 nm,周期性横纹间隔60 nm。此外,有学者认为血清胶原Ⅲ水平升高或可作为Ⅲ型胶原肾小球病的辅助诊断指标[21]。
Ⅲ型胶原肾小球病目前主要以消除水肿、控制血压等对症治疗为主。不同于儿童患者,成人患者可以表现为长期蛋白尿,肾功能正常或缓慢减退,部分患者可进入终末期肾病[22]。肾活检时SCr水平可能是SCr翻倍的独立预测因素[23]。本研究中病例4在肾活检时处于慢性肾脏病5期,在确诊3个月后开始肾脏替代治疗,预后不良;病例1患者应用激素和免疫抑制剂,病例2患者仅应用血管紧张素转化酶抑制剂(ACEI)治疗,经中长期随访,病情都比较稳定。ACEI、激素或免疫抑制剂能否减少患者的蛋白尿,延缓慢性肾脏疾病的进展,保持肾功能的稳定,值得进一步观察。
综上所述,Ⅲ型胶原肾小球病虽然是少见的遗传性疾病,但在成人患者中可散发,且临床表现和病理表现多样,容易误诊,免疫组织化学和电镜检查是确诊的关键。该病尚无特异性治疗方法,ACEI、激素或免疫抑制剂可能对减少患者的蛋白尿、延缓肾功能减退有一定的帮助。
[ 1 ] ARAKAWA M, HUEKI H, SATO M,et al. Idiopathic mesangiodegenerative glomerulonephropathy: a proposal of a new glomerular disease[J].Jpn J Nephrol,1979, 21:914-915.
[ 2 ] VERNIER R L,HOYER J R, MICHAEL A F. The nail-patella syndrome-pathogenesis of the kidney lesion[J].Birth Defect Orig Artic Ser,1974,10(4):57-59.
[ 3 ] DOMBROS N, KATZ A.Nail patella-like renal lesions in the absence of skeletal abnormalities[J]. Am J Kidney Dis,1982,1(4):237-240.
[ 4 ] TAGUCHI T, TAKEBAYASHI S, NISHIMURA M,et al.Nephropathy of nail-patella syndrome[J]. Ultrastruct Pathol,1988,12(2):175-183.
[ 5 ] 陈惠萍, 徐 峰, 黄 倩, 等. 胶原Ⅲ肾病——形态学特点和临床表现[J]. 肾脏病与透析肾移植杂志, 2011, 20(6): 522-529.
[ 6 ] GUBLER M C, DOMMERGUES J P, FOULARD M, et al. Collagen type Ⅲ glomerulopathy: a new type of hereditary nephropathy[J]. Pediatr Nephrol, 1993,7(4):354-360.
[ 7 ] DUGGAL R, NADA R, RAYAT C S, et al. Collagenofibrotic glomerulopathy-a review[J].Clin Kidney J, 2012,5(1):7-12.
[ 8 ] ANITHA A, VANKALAKUNTI M, SIDDINI V, et al.Type Ⅲ collagen disorders: a case report and review of literature[J].Indian J Pathol Microbiol,2016,59(1): 75-77.
[ 9 ] IMBASCIATI E, GHERARDI G, MOROZUMI K, et al.Collagen type Ⅲ glomerulopathy: a new idiopathic glomerular disease[J].Am J Nephrol, 1991,11(5):422-429.
[10] VOGT B A, WYATT R J, BURKE B A, et al. Inherited factor H deficiency and collagen type Ⅲ glomerulopathy[J]. Pediatr Nephrol,1995, 9(1):11-15.
[11] FERREIRA R D, CUSTDIO F B, GUIMARES C S,et al.Collagenofibrotic glomerulopathy: three case reports in Brazil[J]. Diagn Pathol, 2009,4: 33.
[12] ALCHI B, NISHI S, NARITA I, et al. Collagenofibrotic glomerulopathy: clinicopathologic overview of a rare glomerular disease[J]Am J Kidney Dis, 2007, 49(4): 499-506.
[13] TAMURA H, MATSUDA A, KIDOGUCHI N, et al. A family with two sisters with collagenofibrotic glomerulonephropathy[J]. Am J Kidney Dis, 1996, 27(4):588-595
[14] NARUSE K, ITO H, MORIKI T, et al. Mesangial cell activation in the collagenofibrotic glomerulonephropathy. Case report and review of the literature[J].Virchows Arch,1998,433(2):183-188.
[15] Y ASUDA T, IMAI H, NAKAMOTO Y, et al. Collagenofibrotic glomerulopathy: a systemic disease[J]. Am J Kidney Dis,1999,33(1):123-127.
[16] GOTO S, NAKAI K, ITO J, et al.Marked elevation of serum hyaluronan levels in collagenofibrotic glomerulopathy[J].Intern Med,2014,53(16):1801-1804.
[17] KURIEN A A, LARSEN C P, COSSEY L N. Collagenofibrotic glomerulopathy[J]. Clin Kidney J,2015,8(5):543-547.
[18] 李 玲, 邹万忠, 王素霞, 等. Ⅲ型胶原肾小球病的形态学观察[J].中华病理学杂志,2005, 34(7): 385-388.
[19] DONG J, WEI H, HAN M, et al. Collagen type Ⅲ glomerulopathy: a case report and review of 20 cases[J]. Exp Ther Med, 2015,10(4):1445-1449.
[20] FUKAMI K, YAMAGISHI S, MINEZAKI T, et al. First reported case of collagenofibrotic glomerulopathy with a full-house pattern of immune deposits[J]. Clin Nephrol,2014, 81(4): 290-295.
[21] YOSHIDA F, YUZAWA Y, SHIGEMATSU H, et al. Nephrotic syndrome with massive accumulation of type Ⅰ and type Ⅲ collagen in the glomeruli[J].Intern Med, 1993,32(2): 171-176.
[22] 刘海静, 陈 剑, 张 燕, 等. Ⅲ型胶原肾小球病的临床病理学特点[J].中华病理学杂志,2014,43(11): 732-735.
[23] BAO H, CHEN H, ZHU X, et al. Clinical and morphological features of collagen type Ⅲ glomerulopathy: a report of nine cases from a single institution[J].Histopathology, 2015,67(4): 568-576.
[本文编辑] 姬静芳
Clinical and pathological analysis of collagen type Ⅲ glomerualopathy
GONG Shao-min1, LUO Wei-li1, JIN Shi1, LIU Xue-guang2, DING Xiao-Qiang1, LIU Hong1*
1. Department of Nephrology, Zhongshan Hospital, Fudan University, Shanghai Institute of Nephropathy and Dialysis,Shanghai Key Laboratory of Kidney and Blood Purification, Shanghai 200032, China2. Department of Pathology, School of Basic Medical Sciences, Fudan University, Shanghai 200032, China
Objective: To provide basis for the diagnosis and treatment of collagen type Ⅲ glomerualopathy by analyzing the clinical and pathological data of patients.Methods: After searching the data of the patients receiving renal biopsy from March 2007 to June 2015 in nephrology department, the clinical and pathological features of patients who were proven as collagen type Ⅲ glomerualopathy were analyzed.Results: Two male and two female patients aged (45.5±10.7) years old were included. None of the patients were reported to have positive familial history of any kidney diseases. None of them were found to have extra renal involvement. The most common clinical manifestations were proteinuria, hypertension and impaired renal function. Membrane proliferative glomerulonephritis, nodular glomerulosclerosis, focal segmental proliferative and sclerosis glomerulonephritis were found under the light microscopy. One of the patients was accompanied by IgA nephropathy. As for the follow-up results, 1 case with minor declined eGFR and proteinuria less than 0.5 g/24 h remained stable in kidney function and proteinuria, 2 cases responding to glucocorticoids and immunosuppressive agents had improvement in renal function and partial remission of proteinuria, and 1 case developed into end-stage renal disease and
hemodialysis.Conclusions: The sporadic cases of collagen type Ⅲ glomerualopathy may be underestimated in clinical practice, characterized by proteinuria, hypertension, and renal dysfunction. The light microscope pathological features of renal biopsy are various, and they are easy to be missed. Immunohistochemistry and electronic microscopy are essential to confirm the diagnosis.
collagen type Ⅲ glomerualopathy; renal histopathology; treatment
R 692
A
2017-02-24 [接受日期] 2017-03-15
国家中医药管理局中医药科学技术研究专项课题(2016ZX07). Supported by Special Subject of Traditional Chinese Medicine Science and Technology of State Administration of Traditional Chinese Medicine(2016ZX07).
龚劭敏, 博士生,主治医师. E-mail: gong.shaomin@zs-hospital.sh.cn
*通信作者(Corresponding author). Tel: 021-64041990, E-mail:liu.hong@zs-hospital.sh.cn
10.12025/j.issn.1008-6358.2017.20170140
猜你喜欢
天津医科大学学报(2021年4期)2021-08-21 02:14:34
东方考古(2021年0期)2021-07-22 06:26:16
摄影之友(影像视觉)(2020年2期)2021-01-14 05:33:40
肾脏病与透析肾移植杂志(2017年2期)2017-05-09 08:11:55
临床与实验病理学杂志(2017年12期)2017-03-20 04:19:57
安徽医科大学学报(2016年12期)2017-01-15 14:21:58
海峡摄影时报(2016年4期)2016-05-30 10:48:04
医学美学美容·中旬刊(2015年2期)2015-10-21 19:58:27
医学研究杂志(2015年9期)2015-07-01 17:28:00
肾脏病与透析肾移植杂志(2015年5期)2015-06-09 06:49:10
