
基于质谱的定量蛋白质组学技术发展现状
2017-09-16 02:35牟永莹顾培明马博闫文秀王道平潘映红
生物技术通报 2017年9期
牟永莹顾培明马博闫文秀王道平潘映红
(1. 中国农业科学院作物科学研究所 农作物基因资源与基因改良重大科学工程,北京 100081;2. 东北农业大学,哈尔滨 150036)
技术与方法
基于质谱的定量蛋白质组学技术发展现状
牟永莹1,2顾培明1马博1闫文秀1王道平1潘映红1
(1. 中国农业科学院作物科学研究所 农作物基因资源与基因改良重大科学工程,北京 100081;2. 东北农业大学,哈尔滨 150036)
蛋白质组定量分析技术是支撑蛋白质组学研究的关键技术之一,随着蛋白质组定量分析技术的发展,基于质谱的定量蛋白质组学已成为蛋白质组学研究的重要分支。蛋白质组学定量技术可分为非靶向定量和靶向定量两类,靶向定量技术有MRM和PRM模式,非靶向定量技术有非标记定量和体内外标记定量模式,目前使用最多的同位素标记试剂是iTRAQ和TMT。蛋白质组定量技术按数据采集模式还可分DDA和DIA两类。通过对国内外相关文献收集和分析,系统介绍了蛋白质组质谱定量技术的主要特点和发展现状,旨在为生命科学研究者更好地应用定量蛋白质组学技术提供帮助。
定量蛋白质组学;靶向定量;稳定同位素标记;非标记定量;翻译后修饰定量
蛋白质组学(Proteomics)是一门在器官、组织、细胞和亚细胞水平上研究完整蛋白质组表达、翻译后修饰以及蛋白质间相互作用的新兴学科。随着人类及多种模式生物基因组全序列测定工作的完成,蛋白质组学在后基因组时代迅速兴起[1-4]。早期的蛋白质组学研究主要集中在定性分析方面,随着研究的不断深入,提供蛋白质种类和修饰类型等定性信息的蛋白质组学分析技术已经不能满足实际研究需求,因此基于不同原理的蛋白质组定量技术被陆续开发[5-7]。现有的蛋白质组定量分析主要基于双向电泳(Two-dimensional electrophoresis,2-DE)[8,9]和质谱(Mass spectrometry,MS)[10,11]两大类技术,近10年来随着高精度生物质谱技术和数据处理技术的快速发展,基于质谱的蛋白质组定量技术已成为主流的分析手段[12]。本文介绍了目前基于质谱的主要蛋白质组定量技术和一些常用的数据处理软件,以及定量蛋白质组学技术在翻译后修饰蛋白质定量中的应用。
1 基于质谱的定量蛋白质组学技术概况
蛋白质组定量分析在起步阶段主要依赖2-DE技术,通过分析胶图上分离到的蛋白质进行定量,基于质谱的蛋白质组学定量技术则通过对酶切肽段的液相色谱分离和质谱分析完成定量。根据是否对目标蛋白进行定量,基于质谱的蛋白质组学定量技术可分为非靶向定量蛋白质组学(Untargeted quantitative proteomics)和靶向定量蛋白质组学(Targeted quantitative proteomics),其中靶向定量技术包括多重反应监测技术(Multiple reaction monitoring,MRM)和平行反应监测(Parallel reaction monitoring,PRM),非靶向定量技术包括非标记定量和稳定同位素标记定量,稳定同位素标记又可分为多种模式,最值得关注的是等重同位素标记相对和绝对定量(Isobaric tags for relative and absolute quantitation,iTRAQ)和串联质量标签(Tandem mass tags,TMT)技术(图1)。涉及定量蛋白质组学研究的文献近年来显著增长,以proteomics和PRM/MRM/iTRAQ/TMT/Label-free为关键词,分别统计NCBI PubMed “Title/Abstract” 中包含各组关键词的文献数量,发现目前应用最多的是iTRAQ定量和非标定量技术,TMT标记技术和靶向定量技术的应用也快速增长,显示了定量蛋白质组学强劲的发展趋势(图2)。目前质谱定量技术主要采取数据依赖采集模式(Data dependent analysis,DDA),新发展的数据非依赖采集模式(Data independent analysis,DIA)综合了DDA和其他方法的优势,具有更好的分析准确度和动态范围,也值得重点关注。
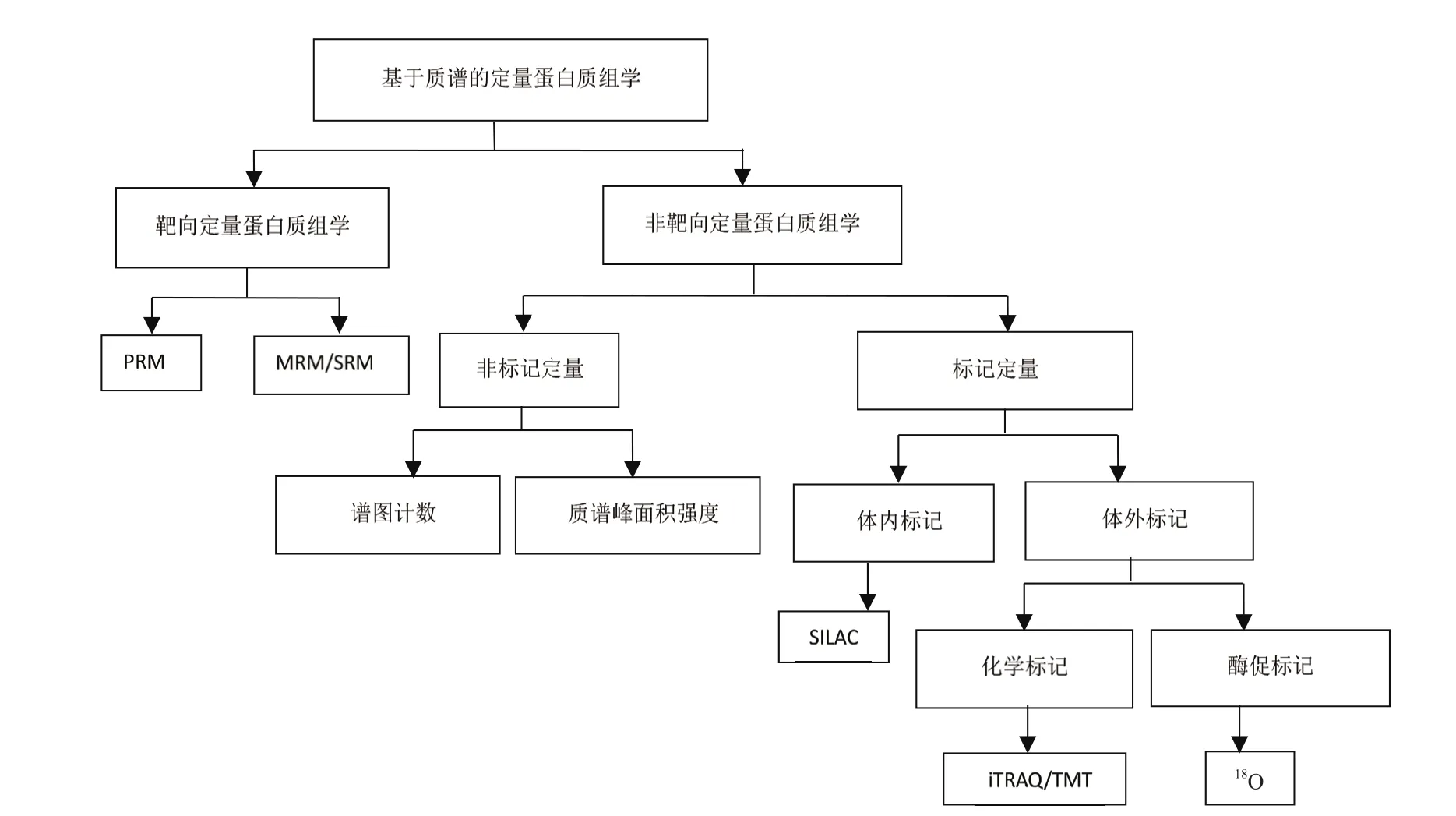
图1 定量蛋白质组学技术分类图
2 非靶向定量蛋白质组学技术
非靶向定量蛋白质组学技术是一种对样品中所有蛋白进行无差别分析的定量技术。根据是否对蛋白质或多肽进行标记,非靶向定量蛋白质组学技术可分为非标记(Label-free)和标记(Stable isotope labeling)定量技术。

图2 2004年-2016年基于质谱的定量蛋白质组学技术相关文献数量变化趋势图
2.1 非标记定量技术
非标记定量蛋白质组学技术主要通过计算蛋白肽段匹配的二级谱图鉴定数目和一级质谱峰面积进行相对定量,使用该技术的优势在于成本低廉和样品制备简单。
2.1.1 基于二级谱图的非标记定量技术 基于二级谱图的非标记定量技术采用匹配肽段的谱图计数(Spectrum counting)实现蛋白定量。质谱分析肽段混合物样品时,某一肽段被鉴定到的几率与其在混合物中的丰度成正比,丰度高的蛋白质被检测到的肽段数和二级谱图数会更多,基于这一原理的方法叫做谱图计数法[13]。Gao等[14]早期发展的肽段鉴定数目技术(Peptide hits technology)即利用不同样品中肽段数目的比值对蛋白质进行相对定量,后期Griffin等[15]整合肽段数目、谱图数目及二级碎片离子强度这3种质谱丰度特征,开发和测试了归一化非标记定量方法(Normalized spectral index)消除了质谱重复测量之间的差异,提高了分析结果的重复性。传统的谱图计数法利用质谱鉴定到的全部肽段进行定量,而部分肽段可能属于两个或多个蛋白共有的非特异性肽段,因此定量的准确性会受到影响。2015年,Zhang等[16]提出使用每个蛋白质的特征肽段进行定量,可有效提高非标记定量的准确性,在样本中加入已知量的标准蛋白还可以进行绝对定量。2.1.2 基于一级谱图的非标记定量技术 基于一级质谱的非标记定量的依据是质谱峰面积强度(Peak area intensity),其原理最早由Chelius等[17]提出并验证,即每条酶解多肽的质谱信号强度与其浓度相关,因此比较一级谱图中的离子信号强度或峰面积,就能确定不同样品中对应蛋白质的相对含量,这类方法被称为离子强度法或信号强度法。Silva等[18]最先发现蛋白质浓度与其所含的3个信号最强肽段的质谱信号平均值呈线性相关,受到Silva的启发,Grossmann等[19]根据类似原理拓展了T3PQ(Top 3 Protein Quantification)非标记定量计算方法,并证明该方法适用于DDA所采集的数据,可用于蛋白质的相对和绝对定量。伴随着质谱技术和软件的发展,基于一级质谱的蛋白质非标记定量技术已得到较广泛的应用,近年来一系列配套的数据处理软件和程序也应运而生,如SAINT-MS1[20],QPROT[21],RIPPER[22]等。
2.2 标记定量技术
标记定量的主要策略是向不同蛋白质或多肽样品中引入具有稳定同位素标记的小分子,通过同位素标记后所产生的质量差来识别肽段的来源。在同一次质谱扫描中化学性质相同的标记肽段离子化效率和碎裂模式也相同,因此比较不同的同位素标记物的信号强度就可以计算出不同样品中蛋白质的相对含量[23]。该方法的优点在于将不同样本混匀后同时进行质谱检测,可以避免样品前处理所带来的定量误差。根据引入同位素标记方式的不同,同位素标记的定量蛋白质组学技术分为体内标记和体外标记两类。
2.2.1 体内标记定量技术 经典的体内标记定量技术是稳定同位素氨基酸细胞培养技术(Stable isotope labeling by amino acids in cell culture,SILAC)[24],该技术的基本原理是将轻、重同位素标记的必需氨基酸(通常为赖氨酸和精氨酸)分别加入到细胞培养基中,经过5-6个倍增周期,细胞内新合成的蛋白质氨基酸几乎完全被稳定同位素标记,根据混合样品中两种同位素标记肽段呈现的峰强度或面积比例即可实现对蛋白质的精确定量。SILAC技术在蛋白层次对样本进行混匀,可以有效避免后续酶解等操作所带来的定量误差,具有标记效率高和定量准确性高的特点,主要缺点是存在同位素标记的精氨酸代谢转换成脯氨酸的现象[25],导致标记效率偏低,定量准确性下降,同时该技术早期只适用于活体培养的细胞,对于医学研究常用的组织、体液等样品则无法应用。为了克服上述缺点,在SILAC的基础上发展出了一些新的技术。Super-SILAC技术[26]将SILAC标记方法培养的细胞作为内标加入到人组织样品中,实现对临床组织样品的定量分析,扩大了SILAC技术的应用范围。最近,Coon研究组将中子编码(Neutron encoding,NeuCode)与SILAC技术结合发展为NeuCode SILAC[27],提高了标记效率,使蛋白质组分析具有良好的动态范围和精度,理论上可达到39重标记。
2.2.2 体外标记定量技术 基于代谢反应的体内标记定量技术存在着耗时长、价格贵等问题,因而发展出一系列体外标记定量技术,其中在样品处理后期进行酶促标记和化学标记是当前研究的重点。
2.2.2.1 酶促标记技术18O酶促标记技术由Fenselau实验室首次应用[28],这种技术通过胰蛋白酶的催化作用将一组样品的肽段C末端16O原子替换成
18O,从而使两组样品产生分子量的差异,通过比较标记肽段和未标记肽段的峰面积,即可对蛋白样本进行定量。该技术具有价格低廉、操作简便等优点,缺点是标记稳定性差且易发生18O-16O回标反应。为了解决这一问题,近年来对该方法进行了一系列完善和改进。Zhao等[29]采用微波加热提高反应速率,使用高浓度还原剂和烷基化试剂将胰蛋白酶灭活,有效提高了18O的标记效率并抑制了18O-16O回标反应。Modzel等[30]提出了一种用18O和二甲氨基偶氮苯甲酰(Dabsyl)双重标记肽段的新方法,该方法廉价高效,且定量可信度高。颜辉[31]也报道在18O标记肽段的C端的同时,用金属标记肽段的N末端,实现双重等重标记,结合MRM技术进行目标蛋白质的绝对定量,可提高定量的准确性。
2.2.2.2 化学标记技术 化学标记技术利用化学反应在蛋白质或肽段上引入同位素基团实现样品标记,是发展最快的一类体外标记定量技术,目前在定量蛋白质组学研究中应用广泛。这类技术依据标记基团和检测方法不同有多种类型,常见的有基于一级质谱的同位素亲和标签(Isotope coded affinity tags,ICAT)技术和二甲基化标记(Dimethyl Labeling)技术,以及基于串级质谱的iTRAQ技术和TMT技术。
基于一级质谱的化学标记技术直接在一级质谱谱图中比较轻重标记样品的峰面积或强度,目前在定量蛋白质组分析领域应用不多。Gygi等[32]发展了一种针对半胱氨酸巯基(-SH)的ICAT试剂,这一试剂由反应基团、连接基团和生物素标签组成,可用于定量比较细胞和组织中球蛋白的表达,使蛋白质组分析更简单、准确和快速。但ICAT技术无法用于不含巯基的蛋白或肽段的定量分析,这一缺点导致其应用范围受限。二甲基化标记技术由Hsu等[33]提出,其原理是利用甲醛和氰基硼氢化钠组合标记肽段所有的活性氨基,具有快速、高效、价格低廉等优点。最近,Wu等[34]将甲醛和氰基硼氢化钠分子上的H用D取代,甲醛上的12C用13C取代,组成五重标记试剂,可极大地提高标记效率。
基于串级质谱的化学标记技术通过比较二级或者三级质谱谱图中不同样品的定量报告离子的峰强度实现定量分析,是目前主流的蛋白质组标记定量技术。等重同位素标记相对和绝对定量技术(iTRAQ)是Ross等[35]发展的一种体外同位素标记的相对定量技术,该技术利用多种同位素试剂标记蛋白多肽N末端或赖氨酸侧链基团,具有通量高、稳定性强、应用范围广等优点,可同时比较最多达八组样品的蛋白表达量。串联质量标签(TMT)试剂[36]与iTRAQ试剂的原理类似,由报告基团、平衡基团和反应基团这3部分组成(图3)。常用的TMT试剂盒有2-plex、6-plex和10-plex三种,前两者分别标记2组和6组样品,10-plex通过使用报告离子质荷比差异为6mDa的中子标签可标记10组样品[37]。目前TMT试剂在蛋白质组学研究中应用也比较多,这类定量技术可以结合其他标记技术和高分辨质谱技术,实现更高通量的蛋白质组学定量分析。例如,Dephoure等[38]将TMT技术与SILAC相结合,实现了18重(chong)(18-plex)标记,再配合高分辨率的质谱仪,可对多种蛋白质进行精确的定量分析。Wojdyla等[39]利用TMT标记和抗体富集方法对半胱氨酸氧化及其逆反应进行组合分析,这一方法替代了现有的半胱氨酸氧化分析法,有助于了解半胱氨酸亚硝基化(S-nitrosylation,SNO)和亚磺酰化(S-sulfenylation,SOH)之间的关系。Zhang[40]还提出了一种增强各种同位素标记数据可比性的方法,可显著提高现有iTRAQ 4-plex、iTRAQ8-plex、TMT6-plex和TMT 10-plex结果的应用价值。

图3 TMT 10-plex和iTRAQ 8-plex构成
传统的iTRAQ与TMT技术均使用二级谱图(MS2)进行蛋白质定量,但由于母离子共筛选(coisolation)和共碎裂(co-fragment)干扰,定量结果的准确度会受到影响。三级质谱(MS3)定量利用高分辨率的多功能质谱选择目标离子进行诱导碰撞解离(Collision-Induced Dissociation,CID),然后从中过滤出信号最强的MS2碎片离子,进一步进行高能碰撞解离(High energy collision dissociation,HCD),利用MS3谱图进行定量,有效降低了母离子共筛选和共碎裂干扰的影响[7]。与MS2定量技术相比,MS3定量的问题是采集速度较慢,离子碎裂效率较低,导致定量蛋白质数目减少,定量灵敏度下降。为解决这一问题,Dayon等[41]提出将气相色谱分离(Gas-phase fractionation,GPF)与MS3分析模式结合,并证明在使用GPF与MS3结合分析TMT标记的胰蛋白酶时,定量的精度、准确度和蛋白质组覆盖率均有所提高。McAlister等[42]提出的MultiNotch MS3定量蛋白质组学技术,采集报告离子数是标准MS3的10倍,增加了报告离子动态范围,降低报告离子的方差,极大提高了定量蛋白数目和准确度。
3 靶向定量蛋白质组学
传统的非靶向蛋白质组学定量技术的重复性、灵敏度和分析效率较低,值得庆幸的是,近年来可弥补上述质谱定量缺陷的靶向定量技术取得了长足的进步[43]。目前文献报道的靶向定量蛋白质组学技术主要有多重反应监测技术(MRM,又称选择反应监测技术Selected reaction monitoring,SRM)和平行反应监测技术(PRM)。
3.1 多重反应监测技术
MRM/SRM技术选择目标蛋白的特定母离子和子离子对,进行质谱分析,最大限度排除干扰离子的影响,显著提高了目标肽段的信噪比,是一种很有前景的高通量靶向蛋白定量技术。该技术具有灵敏度高、准确性好、特异性强的优点,被誉为质谱定量的“金标准”,特别适用于标志蛋白的高通量验证。Dominik等[44]以基于MRM的多重累积方法在人血浆中成功地定量监测了67个心血管疾病的生物标志物,Whiteaker等[45]将SRM与基于抗体的免疫亲和富集相结合实现了复杂样品中目标蛋白的精准定量。MRM技术还可对目标蛋白进行绝对定量,如Gerber等[46]通过向样本中掺入已知浓度的合成同位素标准肽段,实现了对人的分离酶蛋白(Human separase protein)的绝对定量。
3.2 平行反应监测技术
PRM也可在复杂样品中同时对多个目标蛋白进行相对或者绝对定量。与MRM相比,PRM采集目标肽段的高分辨率MS2质谱图后,使用相关软件在ppm级别的质量偏差窗口范围内对定量子离子进行峰面积抽提,可以有效排除背景离子的干扰。Kim等[47]利用该技术分析检测了83个酪氨酸激酶,Ronsein等[48]的研究也证明PRM定量的动态范围较广且精度较高。PRM技术的缺点是待分析肽段的数量过大时,需要精细调整质谱采集参数,否则会极大影响定量数据的准确度。2015年,Gallien等[49]设计了一种内标触发并联反应监测新方法(Internal standard triggered-parallel reaction monitoring,ISPRM),通过添加内标和对采集参数的实时调整来定量内源肽段,在完成大量肽段分析的同时也可使质谱始终保持高性能状态。
4 数据依赖和非依赖采集技术
数据依赖采集技术(DDA)已广泛的用于复杂样品中蛋白质的定性和定量分析,然而DDA一级扫描过程中随机选择肽段进行裂解时总是偏向信号强的肽段,因此易造成低丰度肽段的丢失。随着高分辨率,高扫描速度质谱的出现,数据非依赖采集技术(DIA)越来越受到重视,并入选《Nature Methods》2015年度最值得关注技术。DIA采集数据时,首先将质谱扫描的整个质量轴切割为若干区域,使用四级杆或者离子阱将各个区域的母离子筛选出来,再利用高分辨质谱采集该区域范围内所有母离子的全部碎片离子信息,最后根据DDA的谱图库信息抽提出DIA数据中对应的子离子信息用于最终的定量分析。简单来说,在任意一个洗脱时间点,DDA模式通常按照离子强度由高到低采集母离子的碎片离子信息,采集结果具有一定随机性;DIA模式在整个检测时间内,将整个质量轴切割为固定的几个部分,按照顺序进行检测,所有母离子碎片离子均采集;MRM模式在设定的时间窗口内仅仅对某些特定母离子采集特定或者全部的碎片离子(图4)。与DDA相比,DIA可对样品中所有离子的碎片信息进行无偏向性的数据采集,提升了定量结果的重复性和准确性[50];与MRM/PRM相比,DIA数据采集不受指定目标肽段的限制,可用于未知蛋白和大规模蛋白的定量分析[51]。
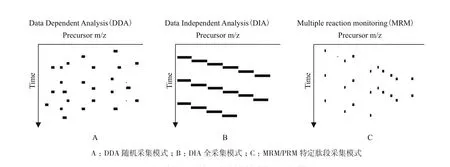
图4 DDA、DIA和MRM的原理比较图
DIA技术是一种高通量的蛋白质组学研究手段,特别适合于临床生物标志物的研究,如Zhong等[52]采用DIA技术筛选出了结核病的潜在血清标志物;Muntel等[53]开发了快速可靠的尿蛋白质组学DIA技术,实现了大规模的尿蛋白质组学研究。在现有的DIA技术中,SWATH(Sequential window acquisition of all theoretical spectra)策略已被较广泛应用,Gillet等[54]使用该技术对酵母全细胞蛋白进行了定量分析;Ortea等[55]利用SWATH-MS和靶向数据提取技术发现了肺腺癌的潜在蛋白质生物标志物。DIA可实现对所有母离子的检测,但选择性相对于DDA或MRM降低了5-10倍,采集的MS2谱图为所有母离子的碎片离子,易造成子离子定量数据的混乱。Egertson等[6]开发的msxDIA技术,可以使得母离子的选择性提高5倍,降低数据处理的复杂度。Tsou等[56]开发的DIA-Umpire软件,首先对DIA数据进行母离子-子离子峰匹配,将DIA数据“转换”为Pseudo-MS/MS MGF 文件,然后使用其他DDA搜库软件进行检索并将该检索结果作为谱图库,最后再使用DIA-umpire软件对DIA数据进行峰面积抽提定量分析,与传统的DIA数据处理模式相比其优势在于研究者无需采集DDA数据用于构建谱图库。毫无疑问,克服了DDA随机性和MRM低通量缺点的DIA/SWATH技术,将成蛋白质组定量和标志蛋白发现与验证的强有力工具。但任何一种技术都不是完美无缺的,需要根据实际情况选择适用的定量技术(表1)。
5 翻译后修饰蛋白定量技术
生物体细胞对蛋白质合成后进行的共价加工即翻译后修饰(Post-translational modification,PTM)与许多重要的生命进程控制相关,因此全面揭示翻译后修饰的发生规律才能理解蛋白质复杂多样的生物功能。蛋白质发生翻译后修饰时其分子质量会发生相应的改变,如发生磷酸化修饰时分子量会增加79.966 Da,通过质谱可对翻译后修饰蛋白质进行精确的定性与定量研究。常见的PTM有磷酸化、乙酰化、泛素化和糖基化翻译后修饰,分析流程基本相同,主要涉及修饰蛋白质或修饰肽段富集,以及修饰位点的定性和定量检测。
5.1 磷酸化修饰蛋白定量
磷酸化是蛋白质翻译后修饰技术最为重要的一种形式。真核生物中大概有一半以上的蛋白质可发生磷酸化修饰,蛋白质磷酸化几乎调节着整个生命活动过程。因此,定量分析内部和外部因子作用下磷酸化蛋白质的分子调控机制对理解生物功能非常重要[57]。磷酸化蛋白或肽段富集目前主要有抗体富集法、固相金属离子亲和色谱法、金属氧化物亲和色谱法、强阴阳离子交换色谱法和亲水相互作用色谱法。Mertins等[58]将iTRAQ标记试剂与磷酸化富集技术结合起来对磷酸化肽段进行定性和定量分析,这种定量方法可进行多个条件或时间点样品间的比较分析,在磷酸化定量蛋白质组分析中得到广泛的应用。Piovesana等[59]将Graphitized carbon black(GCB)与金属氧化物TiO2制备成磁性复合材料(mGCB@TiO2),优化了磷酸化肽段的选择性富集过程。近年来,随着质谱技术的进步,对磷酸化蛋白质组进行规模化分析已成为现实[60]。
5.2 乙酰化修饰蛋白定量
蛋白质的乙酰化修饰是通过乙酰转移酶的作用,在蛋白质赖氨酸残基上添加乙酰基的过程。乙酰化修饰影响着细胞生理的各个方面,如蛋白质翻译、折叠、DNA包装和线粒体代谢等,在染色体结构形成及核内转录调控因子激活方面具有重要作用[61],对其进行定量分析可为代谢类疾病的药物研发提供重要依据[62,63]。将SILAC或iTRAQ标记技术与乙酰化抗体富集技术相结合,即可对细胞样本中的乙酰化修饰进行相对定量。Zhang等[64]将抗体富集技术与高精度的质谱相结合,在大肠杆菌中鉴定到了349个乙酰化蛋白和10 709个乙酰化位点,相对于传统方法分别提升了3倍和8倍,实现了大规模的赖氨酸乙酰化蛋白质的鉴定。
5.3 泛素化修饰蛋白定量
泛素化修饰是指一个或多个泛素分子在一系列酶的作用下与底物蛋白质分子共价结合的翻译后修饰过程[65]。泛素化在蛋白质的代谢调节中起着重要作用,可参与细胞分裂增殖、基因表达、信号传导、炎症免疫等几乎一切生命活动。通过将非标记定量技术与泛素化抗体肽段富集方法相互结合,可以实现对泛素化肽段同时定性和定量的目的。Cai等[66]提出了利用蛋白质序列中氨基酸的理化性质(Physicochemical properties,PCPs)对泛素位点进行预测,建立了6个泛素集对其进行验证,结果表明这一方法与其它方法相比具有很大优势。Akimo等[67]描述了一个将特异性的泛素结合结构域(Ubiquitin binding domains,UBDs)与SILAC技术相结合,以

表1 定量蛋白质组学主要技术特点与应用
6 定量蛋白质组学数据处理软件
质谱鉴定是定量蛋白质组学研究的关键步骤之一,其核心在于对质谱数据的解析。目前常用的数据库检索软件包括MaxQuant、Mascot、Sequest、X!Tandem等,依赖于谱图库的检索软件有Pepitome、SpectraST等,其他相关软件有Skyline,Spectronaut等。
MaxQuant是常用的标记和非标记定量蛋白质组学数据分析平台,该平台广泛使用的集成搜索引擎Andromeda可以一种简单的分析工作流程实现大规模数据集的分析[73]。Tyanova等[74]开发了优化MaxQuant报告结果的工具Maxreport,以帮助用户更好的理解和分享数据。Mascot和Sequest是利用分子序列数据检索的方法来鉴定样本中蛋白质组成的经典软件,Brosch等[75]提出了对数据搜索结果再评分的算法Mascot Percolator,允许使用多重替代碎裂技术直接比较鉴定肽段,可作为独立工具或集成到现有的数据分析流程中[76]。X!Tandem是一个在蛋白质鉴定过程中将质谱与肽序列匹配起来的开源软件,用户可以按照自己的需求对其源代码进行修实现对泛素相关的细胞信号通路进行选择性定量的蛋白质组学研究策略。Meng等[68]利用4个相同的泛素相关结构域(Ubiquitin-associated domains,UBA)构建了GST-qUBAs诱饵蛋白,并且利用该诱饵蛋白和两步亲和富集法完成了水稻等植物泛素化修饰蛋白的富集和分析。
5.4 糖基化修饰蛋白定量
糖基化是蛋白质或脂质在酶的催化下结合上不同类型糖基的过程。糖基化修饰影响蛋白质的空间结构和生物活性,是最重要、最复杂的翻译后修饰过程之一,在蛋白质折叠、定位和转运以及细胞生长、病毒复制、免疫保护等生物过程中起着重要的作用[69-70]。糖基化蛋白富集方法主要有氧化石墨烯固定化凝集素法、亲水相互作用色谱法、代谢标签法、化学酶促标记法和抗体富集法等,分析的难点主要在于质谱鉴定方面,即特异性糖链结构的解析,但近年来已有较多研究结果,如Hashii等[71]利用氚标记定量分析了小鼠肾脏糖基化的变化;Ahn等[72]将MRM与Aleuria aurantia lectin(AAL)富集结合,用于肝癌血浆中异常蛋白质糖基化定量。改,如Yang等[77]开发的Self-boosted Percolator,处理X!Tandem搜索结果时可提高肽段鉴定能力。
Pepitome是一种新的依赖于质谱谱图库的搜索引擎,该软件采用统计方法进行评分,可靠性较高[78]。SpectraST是另一种功能全、通量高的开源MS2谱图检索软件,适用于序列测定和靶向蛋白质组学分析[79]。
Skyline是一款由美国华盛顿大学MacCoss实验室开发的开源文档编辑器,支持使用和创建各种资源的MS2谱库,可依据之前观察到的数据来选择SRM过滤器并验证结果,常用于靶向蛋白质组学方法建立和定量数据分析[80]。Spectronaut是由瑞士蛋白质组学公司Biognosys AG开发的可快速、准确分析处理DIA或SWATH数据的软件,具有强大的峰选择算法,可自动质控并修正干扰[81]。
7 小结
定量蛋白质组学在现代生物学和医学研究中发挥着重要作用。目前主要应用的定量蛋白质组学技术有体内标记技术SILAC、基于串级质谱的同位素标记技术TMT和iTRAQ、非标记定量技术、目标离子检测技术MRM/PRM,以及数据非依赖采集技术DIA/SWATH,其中MRM/PRM和DIA/SWATH是特别值得关注的定量蛋白质组学新技术。此外,MS3定量技术解决了同重同位素标记时共洗脱肽段干扰的问题,有望成为蛋白质组学定量的新工具。翻译后修饰蛋白质组的定量分析涉及多种富集方法和质谱技术,目前正处于迅速发展阶段,有望成为定量蛋白质组学研究的一个重要新领域。未来定量蛋白质组学技术的发展方向是进一步提高通量、准确度、稳定性和自动化程度。数据处理软件也是定量蛋白质组学技术的关键组件,有待在易用性、可靠性和计算效率方面进一步发展。
致谢:
中国农业科学院科技创新工程(作物分子标记技术及其应用科研创新团队)和国家重点研发计划(编号:2016YFD0101005)对项目研究给予了经费支持;徐江研究员和郑兆彬工程师对本文的撰写也给予了热情帮助,特致谢意。
[1] Adams MD, Celniker SE, Holt RA, et al. The genome sequence of Drosophila melanogaster[J]. Science, 2000, 287(5461):2185-2195.
[2] Gregory SG, Barlow KF, Mclay KE, et al. The DNA sequence and biological annotation of human chromosome 1[J]. Nature, 2006, 441(7091):315-321.
[3] Muzny DM, Scherer SE, Kaul R, et al. The DNA sequence, annotation and analysis of human chromosome 3[J]. Nature, 2006, 440(7088):1194-1198.
[4] Yu J, Hu SN, Wang J, et al. A draft sequence of the rice genome(Oryza sativa L. ssp indica)[J]. Science, 2002, 296(5565):79-92.
[5] Kruger M, Moser M, Ussar S, et al. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function[J]. Cell, 2008, 134(2):353-364.
[6] Egertson JD, Kuehn A, Merrihew GE, et al. Multiplexed MS/MS for improved data-independent acquisition[J]. Nature Methods, 2013, 10(8):744-746.
[7] Ting L, Rad R, Gygi SP, et al. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics[J]. Nature Methods, 2011, 8(11):937-940.
[8] Marouga R, David S, Hawkins E. The development of the DIGE system:2D fluorescence difference gel analysis technology[J]. Analytical and Bioanalytical Chemistry, 2005, 382(3):669-678.
[9] Wu WW, Wang GH, Baek SJ, et al. Comparative study of three proteomic quantitative methods, DIGE, cICAT, and iTRAQ, using 2D gel- or LC-MALDI TOF/TOF[J]. Journal of Proteome Research, 2006, 5(3):651-658.
[10] Boersema PJ, Raijmakers R, Lemeer S, et al. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics[J]. Nature Protocols, 2009, 4(4):484-494.
[11] Levin Y, Schwarz E, Wang L, et al. Label-free LC-MS/MS quantitative proteomics for large-scale biomarker discovery in complex samples[J]. Journal of Separation Science, 2007, 30(14):2198-2203.
[12] Chahrour O, Cobice D, Malone J. Stable isotope labelling methods in mass spectrometry-based quantitative-proteomics[J]. Journal of Pharmaceutical and Biomedical Analysis, 2015, 113:2-20.
[13] Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology[J]. Nature Biotechnology, 2001, 19(3):242-247.
[14] Gao J, Friedrichs MS, Dongre AR, et al. Guidelines for the routine application of the peptide hits technique[J]. J Am Soc Mass Spectrom, 2005, 16(8):1231-1238.
[15] Griffin NM, Yu J, Long F, et al. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis[J]. Nat Biotechnol, 2010, 28(1):83-89.
[16] Zhang Y, Wen Z, Washburn MP, et al. Improving label-free quantitative proteomics strategies by distributing shared peptides and stabilizing variance[J]. Analytical Chemistry, 2015, 87(9):4749-4756.
[17] Chelius D, Bondarenko, PV. Quantitative profiling of proteins in complex mixtures using liquid chromatography and mass spectrometry[J]. J Proteome Res, 2002, 1(4):317-323.
[18] Silva JC, Gorenstein MV, Li GZ, et al. Absolute quantification of proteins by LCMSE-A virtue of parallel MS acquisition[J]. Molecular & Cellular Proteomics, 2006, 5(1):144-156.
[19] Grossmann J, Roschitzki B, Panse C, et al. Implementation and evaluation of relative and absolute quantification in shotgun proteomics with label-free methods[J]. Journal of Proteomics, 2010, 73(9):1740-1746.
[20] Choi H, Glatter T, Gstaiger M, et al. SAINT-MS1:Proteinprotein interaction scoring using label-free intensity data in affinity purification-mass spectrometry experiments[J]. Journal of Proteome Research, 2012, 11(4):2619-2624.
[21] Choi H, Kim S, Fermin D, et al. QPROT:Statistical method for testing differential expression using protein-level intensity data in label-free quantitative proteomics[J]. J Proteomics, 2015, 129:121-126.
[22] Van Riper SK, Higgins L, Carlis JV, et al. RIPPER:a framework for MS1 only metabolomics and proteomics label-free relative quantification[J]. Bioinformatics, 2016, 32(13):2035-2037.
[23] 钱小红. 定量蛋白质组学分析方法[J]. 色谱, 2013(8):719-723.
[24] Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics[J]. Molecular & Cellular Proteomics, 2002, 1(5):376-386.
[25] Bicho CC, Alves FD, Chen ZA, et al. A genetic engineering solutionto the “arginine conversion problem” in stable isotope labeling by amino acids in cell culture(SILAC)[J]. Molecular & Cellular Proteomics, 2010, 9(7):1567-1577.
[26] Neubert TA, Tempst P. Super-SILAC for tumors and tissues[J]. Nat Methods, 2010, 7(5):361-362.
[27] Hebert AS, Merrill AE, Bailey DJ, et al. Neutron-encoded mass signatures for multiplexed proteome quantification[J]. Nat Methods, 2013, 10(4):332-334.
[28] Xudong Yao AF, Javier Ramirez, Plamen A. Demirev, and Catherine Fenselau. Proteolytic 18O labeling for comparative proteomics:model studies with two serotypes of adenovirus[J]. Analchem, 2001, 73(13):7.
[29] Zhao Y, Jia W, Sun W, et al. Combination of improved18O incorporation and multiple reaction monitoring:a universal strategy for absolute quantitative verification of serum candidate biomarkers of liver cancer[J]. J Proteome Res, 2010, 9(6):3319-3327.
[30] Modzel M, Plociennik H, Kielmas M, et al. A synthesis of new, bilabeled peptides for quantitative proteomics[J]. J Proteomics, 2015, 115:1-7.
[31] 颜辉. 金属标记结合生物质谱的蛋白质组相对定量及绝对定量新方法研究[D]. 合肥:安徽医科大学, 2014.
[32] Gygi SP, Rist B, Gerber SA, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. [J]. Nat Biotechnol, 1999, 17(10):6.
[33] Hsu J L, Huang SY, Chow NH, et al. Stable-isotope dimethyl labeling for quantitative proteomics[J]. Analytical Chemistry, 2003, 75(24):6843-6852.
[34] Wu Y, Wang F, Liu Z, et al. Five-plex isotope dimethyl labeling for quantitative proteomics[J]. Chem Commun(Camb), 2014, 50(14):1708-1710.
[35] Ross PL, Huang YN, Marchese JN, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents[J]. Mol Cell Proteomics, 2004, 3(12):1154-1169.
[36] Andrew Thompson J, 1rgen Schagunter KK, Stefan Kienle, Josef Schwarz, , 1nter Schmidt TN, And Christian Hamon. Tandem mass tags:A novel quantification strategy for comparative analysis of complex protein mixtures by MS MS[J]. Analchem, 2003, 75(8):10.
[37] Werner T, Sweetman G, Savitski MF, et al. Ion Coalescence of neutron encoded TMT 10-plex reporter Ions[J]. Analytical Chemistry, 2014, 86(7):3594-3601.
[38] Dephoure N, Gygi SP. Hyperplexing:a method for higher-order multiplexed quantitative proteomics provides a map of the dynamic response to rapamycin in yeast[J]. Sci Signal, 2012, 5(217):9.
[39] Wojdyla K, Williamson J, Roepstorff P, et al. The SNO/SOH TMT strategy for combinatorial analysis of reversible cysteine oxidations[J]. Journal of Proteomics, 2015, 113:415-434.
[40] Zhang Z, Yang XY, Mirokhin YA, et al. Interconversion of peptide mass spectral libraries derivatized with iTRAQ or TMT labels[J]. Journal of Proteome Research, 2016, 15(9):3180-3187.
[41] Dayon L, Sonderegger B, Kussmann M. Combination of gasphase fractionation and MS3acquisition modes for relative protein quantification with isobaric tagging[J]. Journal of Proteome Research, 2012, 11(10):5081-5089.
[42] Mcalister GC, Nusinow DP, Jedrychowski MP, et al. MultiNotch MS3enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes[J]. Analytical Chemistry, 2014, 86(14):7150-7158.
[43] Searle BC, Egertson JD, Bollinger JG, et al. Using data independent acquisition(DIA)to model high-responding peptides for targeted proteomics experiments[J]. Molecular & Cellular Proteomics, 2015, 14(9):2331-2340.
[44] Domanski D, Percy AJ, Yang J, et al. MRM-based multiplexed quantitation of 67 putative cardiovascular disease biomarkers in human plasma[J]. Proteomics, 2012, 12(8):1222-1243.
[45] Whiteaker JR, Zhao L, Anderson L, et al. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometrybased quantification of protein biomarkers[J]. Molecular & Cellular Proteomics, 2010, 9(1):184-196.
[46] Gerber SA, Rush J, Stemman O, et al. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS[J]. Proceedings of the National Academy of Sciences of the United States of America, 2003, 100(12):6940-6945.
[47] Kim HJ, Lin D, Lee HJ, et al. Quantitative profiling of protein tyrosine kinases in human cancer cell lines by multiplexed parallel reaction monitoring assays[J]. Molecular & Cellular Proteomics,2016, 15(2):682-691.
[48] Ronsein GE, Pamir N, Von Haller PD, et al. Parallel reaction monitoring(PRM)and selected reaction monitoring(SRM)exhibit comparable linearity, dynamic range and precision for targeted quantitative HDL proteomics[J]. Journal of Proteomics, 2015, 113:388-399.
[49] Gallien S, Kim SY, Domon B. Large-Scale targeted proteomics using internal standard triggered-parallel reaction monitoring(ISPRM)[J]. Molecular & Cellular Proteomics, 2015, 14(6):1630-1644.
[50] Law KP, Lim YP. Recent advances in mass spectrometry:data independent analysis and hyper reaction monitoring[J]. Expert Review of Proteomics, 2013, 10(6):551-566.
[51] Egertson JD, Maclean B, Johnson R, et al. Multiplexed peptide analysis using data-independent acquisition and Skyline[J]. Nature Protocols, 2015, 10(6):887-903.
[52] Zhong LJ, Li Y, Tian HF, et al. Data-independent acquisition strategy for the serum proteomics of tuberculosis[J]. International Journal of Clinical and Experimental Pathology, 2017, 10(2):1172-1185.
[53] Muntel J, Xuan Y, Berger ST, et al. Advancing urinary protein biomarker discovery by data-independent acquisition on a quadrupole-orbitrap mass spectrometer[J]. Journal of Proteome Research, 2015, 14(11):4752-4762.
[54] Gillet LC, Navarro P, Tate S, et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition:A new concept for consistent and accurate proteome analysis[J]. Molecular & Cellular Proteomics, 2012, 11(6):17.
[55] Ortea I, Rodriguez-Ariza A, Chicano-Galvez E, et al. Discovery of potential protein biomarkers of lung adenocarcinoma in bronchoalveolar lavage fluid by SWATH MS data-independent acquisition and targeted data extraction[J]. Journal of Proteomics, 2016, 138:106-114.
[56] Tsou CC, Avtonomov D, Larsen B, et al. DIA-Umpire:comprehensive computational framework for data-independent acquisition proteomics[J]. Nature Methods, 2015, 12(3):258-264.
[57] 甄艳李, 施季森. 磷酸化蛋白质组定量研究策略. [J]. 分子植物育种, 2014, 12(3):7.
[58] Mertins P, Udeshi ND, Clauser KR, et al. iTRAQ labeling is superior to mTRAQ for quantitative global proteomics and phosphoproteomics[J]. Molecular & Cellular Proteomics, 2012, 11(6):12.
[59] Piovesana S, Capriotti AL, Cavaliere C, et al. New magnetic graphitized carbon black TiO2 composite for phosphopeptide selective enrichment in shotgun phosphoproteomics[J]. Analytical Chemistry, 2016, 88(24):12043-12050.
[60] Song C, Ye M, Liu Z, et al. Systematic Analysis of protein phosphorylation networks from phosphoproteomic data[J]. Molecular & Cellular Proteomics, 2012, 11(10):1070-1083.
[61] 阮班军, 代鹏, 王伟, 等. 蛋白质翻译后修饰研究进展 [J].中国细胞生物学学报, 2014, 36(7):1027-1037.
[62] Scholz C, Weinert BT, Wagner SA, et al. Acetylation site specificities of lysine deacetylase inhibitors in human cells[J]. Nature Biotechnology, 2015, 33(4):415-136.
[63] Yang L, Vaitheesvaran B, Hartil K, et al. The Fasted/Fed mouse metabolic acetylome:N6-acetylation differences suggest acetylation coordinates organ-specific fuel switching[J]. Journal of Proteome Research, 2011, 10(9):4134-4149.
[64] Zhang K, Zheng S, Yang JS, et al. Comprehensive profiling of protein lysine acetylation in Escherichia coli[J]. Journal of Proteome Research, 2013, 12(2):844-851.
[65] Lu L, Li D, He FC. Bioinformatics advances in protein ubiquitination[J]. Hereditas(Beijing), 2013, 35(1):17-26.
[66] Cai B, Jiang X. Computational methods for ubiquitination site prediction using physicochemical properties of protein sequences[J]. Bmc Bioinformatics, 2016, 17:116.
[67] Akimov V, Rigbolt KTG, Nielsen MM, et al. Characterization of ubiquitination dependent dynamics in growth factor receptor signaling by quantitative proteomics[J]. Molecular Biosystems, 2011, 7(12):3223-3233.
[68] Meng Q, Rao L, Pan Y. Enrichment and analysis of rice seedling ubiquitin-related proteins using four UBA domains(GST-qUBAs)[J]. Plant Science An International Journal of Experimental Plant Biology, 2014, 229:172-180.
[69] 包慧敏, 谢力琦, 陆豪杰. 糖蛋白质组学中基于化学反应的富集方法研究进展[J]. 色谱, 2016(12):1145-1153.
[70] 张伟. 基于生物质谱的蛋白质N-糖基化定性与定量新技术[D]. 上海:复旦大学, 2012.
[71] Hashii N, Kawasaki N, Itoh S, et al. Alteration of N-glycosylationin the kidney in a mouse model of systemic lupus erythematosus:relative quantification of N-glycans using an isotope-tagging method[J]. Immunology, 2009, 126(3):336-345.
[72] Ahn YH, Shin PM, Kim YS, et al. Quantitative analysis of aberrant protein glycosylation in liver cancer plasma by AAL-enrichment and MRM mass spectrometry[J]. Analyst, 2013, 138(21):6454-6462.
[73] Cox J, Neuhauser N, Michalski A, et al. Andromeda:a peptide search engine integrated into the MaxQuant environment[J]. Journal of Proteome Research, 2011, 10(4):1794-1805.
[74] Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics[J]. Nature Protocols, 2016, 11(12):2301-2319.
[75] Brosch M, Yu L, Hubbard T, et al. Accurate and sensitive peptide identification with mascot percolator[J]. Journal of Proteome Research, 2009, 8(6):3176-3181.
[76] Wright JC, Collins MO, Yu L, et al. Enhanced peptide identification by electron transfer dissociation using an improved mascot percolator[J]. Molecular & Cellular Proteomics, 2012, 11(8):478-491.
[77] Yang PY, Ma J, Wang PH, et al. Improving X! Tandem on peptide identification from mass spectrometry by self-boosted percolator[J]. Ieee-Acm Transactions on Computational Biology and Bioinformatics, 2012, 9(5):1273-1280.
[78] Dasari S, Chambers MC, Martinez MA, et al. Pepitome:evaluating improved spectral library search for identification complementarity and quality assessment[J]. Journal of Proteome Research, 2012, 11(3):1686-1695.
[79] Lam H, Deutsch EW, Eddes JS, et al. Development and validation of a spectral library searching method for peptide identification from MS/MS[J]. Proteomics, 2007, 7(5):655-667.
[80] Maclean B, Tomazela DM, Shulman N, et al. Skyline:an open source document editor for creating and analyzing targeted proteomics experiments[J]. Bioinformatics, 2010, 26(7):966-968.
[81] Bruderer R, Bernhardt OM, Gandhi T, et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues[J]. Molecular & Cellular Proteomics, 2015, 14(5):1400-1410.
[82] Mann M. Functional and quantitative proteomics using SILAC[J]. Nature Reviews Molecular Cell Biology, 2006, 7(12):952-958.
[83] Werner T, Becher I, Sweetman G, et al. High-resolution enabled TMT 8-plexing[J]. Analytical Chemistry, 2012, 84(16):7188-7194.
[84] Pichler P, Koecher T, Holzmann J, et al. Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-plex compared to TMT 6-plex and iTRAQ 8-plex on LTQ orbitrap[J]. Analytical Chemistry, 2010, 82(15):6549-6558.
[85] Neilson KA, Ali NA, Muralidharan S, et al. Less label, more free:Approaches in label-free quantitative mass spectrometry[J]. Proteomics, 2011, 11(4):535-553.
[86] Collins BC, Gillet LC, Rosenberger G, et al. Quantifying protein interaction dynamics by SWATH mass spectrometry:application to the 14-3-3 system[J]. Nature Methods, 2013, 10(12):1246-1253.
(责任编辑 朱琳峰)
Advancements in Quantitative Proteomics Technologies Based on Mass Spectrometry
MU Yong-ying1,2GU Pei-ming1MA Bo1YAN Wen-xiu1WANG Dao-ping1PAN Ying-hong1
(1. Institute of Crop Science,Chinese Academy of Agricultural Sciences,Beijing 100081;2. Northeast Agricultural University,Harbin 150036)
Quantitative proteomic analysis is one of the key technologies in proteomics research. With the development of quantitative techniques,quantitative proteomics based on mass spectrometry has become an important branch of proteomics research. Quantitative proteomics technology is generally classified into two categories,untargeted quantitative proteomics and targeted quantitative proteomics. Targeted quantitative technology includes MRM and PRM models,and untargeted quantitative technology includes label-free and in vivo or vitro labeling. The most commonly used isotope labeling reagents are iTRAQ and TMT. According to the data collection model,the quantitative technology can be grouped into DDA and DIA. Through the collection and analysis of relevant literature,this article systematically introduces the main characteristics and current situation of quantitative proteomics based on mass spectrometry,aiming at providing help for the researchers on life science to better apply the quantitative proteomics technologies.
quantitative proteomics;target quantitative;stable isotope labeling;label-free;post-translational modification
10.13560/j.cnki.biotech.bull.1985.2017-0343
2017-04-28
国家重点研发计划(2016YFD0101005),中国农业科学院科技创新工程
牟永莹,女,硕士研究生,研究方向:蛋白质组学;E-mail:mu_yongying@163.com
潘映红,男,研究员,研究方向:蛋白质组学;E-mail:panyinghong@caas.cn
猜你喜欢
食品安全导刊(2021年20期)2021-08-30
世界科学技术-中医药现代化(2020年2期)2020-07-25
国际口腔医学杂志(2019年3期)2019-05-31
中成药(2018年12期)2018-12-29
天然产物研究与开发(2018年2期)2018-04-04
中成药(2017年6期)2017-06-13
当代化工研究(2016年5期)2016-03-20
医学研究杂志(2015年11期)2015-06-10
医学研究杂志(2015年4期)2015-06-10
特产研究(2014年4期)2014-04-10
