
Spreading depression and focal venous cerebral ischemia enhance cortical neurogenesis
2017-09-04 07:27RyoTamakiSamuelIgeOrieBeatAlessandriOliverKempskiAxelHeimann
中国神经再生研究(英文版) 2017年8期
Ryo Tamaki, Samuel Ige Orie, Beat Alessandri Oliver Kempski Axel Heimann
1 Department of Neurosurgery, Nara Medical University, Nara, Japan
2 University Medical Center of the Johannes Gutenberg-University of Mainz, Institute for Neurosurgical Pathophysiology, Mainz, Germany
Spreading depression and focal venous cerebral ischemia enhance cortical neurogenesis
Ryo Tamaki1,#, Samuel Ige Orie2,#, Beat Alessandri2, Oliver Kempski2, Axel Heimann2,*
1 Department of Neurosurgery, Nara Medical University, Nara, Japan
2 University Medical Center of the Johannes Gutenberg-University of Mainz, Institute for Neurosurgical Pathophysiology, Mainz, Germany
How to cite this article:Tamaki R, Orie SI, Alessandri B, Kempski O, Heimann A (2017) Spreading depression and focal venous cerebral ischemia enhance cortical neurogenesis. Neural Regen Res 12(8):1278-1286.
Endogenous neurogenesis can arise from a variety of physiological stimuli including exercise, learning, or“enriched environment” as well as pathological conditions such as ischemia, epilepsy or cortical spreading depression. Whether all these conditions use a common trigger to set of f endogenous neurogenesis is yet unclear. We hypothesized that cortical spreading depression (CSD) induces neurogenesis in the cerebral cortex and dentate gyrus after cerebral venous ischemia. Forty-two Wistar rats alternatively underwent sham operation (Sham), induction of ten CSDs or venous ischemia provokedviaocclusion of two adjacent superf i cial cortical vein followed by ten induced CSDs (CSD + 2-VO). As an additional control, 15 naïve rats
nerve regeneration; cortical spreading depression; two-vein occlusion; adult neurogenesis; stem cells; cerebral cortex; neural precursor cells; neuron; penumbra; neural regeneration
Introduction
The dogma of the brain’s inability to replace lost neuronal cells has been undeniably refuted. Today at least two regions of the adult brain are known to retain the ability to continuously produce neuronal cells throughout the life. These“neurogenic” regions are the subventricular zone (SVZ) of the lateral ventricle and the subgranular zone (SGZ) of the hippocampal formation (Lois and Alvarez-Buylla, 1994; Cooper-Kuhn and Kuhn, 2002; Carleton et al., 2003).ere is also growing evidence suggesting the presence of neural stem cells in other “non-neurogenic” regions of the brain to include the cerebral cortex, caudate nucleus, and the striatum (Kaplan, 1981; Gould et al., 1999b; Bernier et al., 2002). However, unlike the SVZ and SGZ, comparatively very low levels of neurogenesis exist in these latter regions under physiological conditions.
Transient cortical spreading depression (CSD) is a wave of sustained neuronal depolarization moving through intact brain tissues at the speed of 2–5 mm/min.is phenomenon fi rst described by Leão (1944) can be triggered by increased extracellular potassium following electrical, mechanical or chemical stimuli and is associated with “periinfarct depolarizations.” In clinical and experimental settings, CSD can be induced through the application of potassium chloride (KCl) into the brain.e ef f ects of CSD are presumed to be favorable to brain cells and neuroprotective due to its preconditioning ef f ects, upregulation of trophic substances such as brain-derived neurotrophic factor (BDNF) and protection against subsequent focal brain ischemia (Kokaia, 1993; Matsushima et al., 1998; Yanamoto et al., 2005). However, other studies also suggest that CSD due to increased adenosine triphosphate (ATP) consumption (negative energy balance) and ‘inverse’ vascular coupling may well contribute to increased tissue hypoxia as well as secondary injury in stroke and traumatic brain injury (Nedergaard and Hansen, 1988; Takano et al., 2007).
Based on our previous findings on the pathophysiological processes involved in cerebral ischemia using a rat twovein occlusion model (Nakase et al., 1996, 1997; Heimann et al., 2003), cerebral venous occlusion as a result of relative low fl ow area compared to the ischemic core causes a slowly developing tissue infarction that leads to local tissue demise (Kempski et al., 1999). Unlike the arterial occlusion, the penumbra area after venous occlusion is sustained over a longer period of time. In combination with CSD, the damaging ef f ect on tissue is more severe (Otsuka et al., 2000). Both CSD and cerebral ischemia individually have been reported to augment the response of adult stem cellsin vivo(Tamura et al., 2004; Yanamoto et al., 2005).e aim of this study is therefore to clarify whether CSD alone versus CSD in combination with cerebral venous ischemia is able to trigger neurogenesis in the cerebral cortex and dentate gyrus (DG).
Materials and Methods
Animals
This study is approved by the Landesuntersuchungsamt Rheinland-Pfalz (approval No. AZ: 177-07/051-16), and was performed in accordance with the German animal protection law. All ef f orts were made to minimize the number and suf f ering of animals used in this experiment.
Forty-two male Wistar rats (7–9 weeks old, Charles Rivers, Germany), weighing 315–359 g, were randomized into three groups: Sham (n= 14), CSD (n= 14), and CSD plus two-vein occlusion (CSD + 2-VO;n= 14). A 2-VO group without additional CSD induction was consciously abandoned because of its important variability of spontaneously occurring CSD which go along with altering infarction volume causing problems in statistical and interpretation of pathophysiological pathways (Otsuka et al., 2000). Seven animals in each experimental group were observed for either 9 or 28 days according to their survival time. In order to assess the rate of neurogenesis under physiological conditions two additional groups of naïve animals received 7 days of BrdU treatment and were sacrif i ced aer 9 (n= 7) and 28 days (n= 8), respectively. Animals were housed in individual cages and allowed free access to food and waterad libitumprior to and aer surgery.
Animal preparation
Animals were premedicated with 1 mg atropine sulfate and anesthesia was performed by intraperitoneal injection of chloral hydrate (36 mg/100 g body weight). Rats were intubated and mechanically ventilated with 30% oxygen under controlled end respiratory PCO2(Artema MM206C; Heyer, Sweden) using a rodent ventilator (Model 683; Harvard Apparatus, MA, USA). Rectal temperature was maintained at 37°Cviaa feedback-controlled heating pad (Harvard Apparatus, MA, USA).e tail artery was cannulated using a polyethylene catheter (outer diameter 0.96 mm) to measure arterial blood pressure (MABP; Gould transducer 134615-50), and to monitor blood gases, electrolytes, glucose, hematocrit and pH levels (ABL System 612/EML6, Radiometer, Denmark) during operation.e femoral vein was catheterized for drug administration. Rats were placed in a stereotactic frame (Stoelting, Wood Dale, IL, USA) a lecranial window was drilled under an operating microscope (OP-Microscope; Zeiss, Wetzlar, Germany) to access the brain. To avoid thermal injury, the tip of the drill was continuously cooled with physiological saline during craniotomy. As described previously (Nakase et al., 1996, 1997), regional cerebral blood fl ow (rCBF) was measured using laser Doppler (LD) scanning (Model BPM 403a; Vasomedics, St. Paul, MN, USA) with a 0.8-mm needle probe. Flow is expressed in LD units. A micropipette (GB150F10, Science Products GmbH Hoeim, Germany pulled by Micropipette Puller P-87, Navato, CA, USA) was inserted into the cerebral cortex for application of KCl. Baseline values were taken 90 minutes aer insertion, before initiation of venous ischemia.
Cortical vein occlusion by photochemical thrombosis
Two adjacent superficial cortical veins connecting into the superior sagittal sinus were occluded using Rose Bengal dye (Sigma Chemical Co., St. Louis, MO, USA) in combination with fiberoptic illumination (100-W mercury lamp [6,500–7,500 lx, 540 nm]) connected to a 200-µm fiber. Only animals which presented similar anatomy (i.e., two prominent adjacent cortical veins connecting into the superior sagittal sinus) were used in this study.e diameter of occluded veins was approximately 80–100 µm. Rose Bengal at the dose of 50 mg/kg was slowly injected intra¬venously; target veins were then selectively illuminated using a micromanipulator-assisted fi ber-optic light guide with care taken to avoid illumination of nearby tissue and other blood vessels. Target veins were illuminated for 10 minutes. Prior to the illumination of the second target vein, half of the initial Rose Bengal dosage was additionally administered intravenously before the illumination was undertaken. Occlusion of veins was conf i rmed with the laser Doppler system detecting acutely decreased blood fl ow at the site of the veins and its surrounding tissues.
Induction of cortical spreading depression
All animals randomly assigned to receive CSD or CSD + 2-VO occlusion were subjected to 10 cortical spreading depressions induced via the intracerebral application of 2 µL of a 150 mM potassium chloride (KCl) solution by a glass micropipette and a microinjection pump (CMA/100; Carnegie Medicine, Stockholm, Sweden), with 7-minute intervals between each injection, and administration procedures lasting for 70 minutes overall. Rats in the CSD group were subjected to KCl administration without previous 2-VO, while rats in the combined group received 2-VO before KCl administration. Tissue impedance as an indicator for cell swelling during CSD was measured continuously using two impedance electrodes (0.4–0.5 mm depth, 3 mm from occluded vein, stainless steel wires, outside diameter 0.5 mm) covered with polyurethan insulating sheath - except for its exposed tips (outer diameter 0.3 mm). Impedance measurements were recorded at 1 kHz (1 µA, 10 mV, bias-free) continuously throughout the course of the experiment using a precision LCR monitor (4284A; Hewlett-Packard, Hewlett-Packard, Salt Lake City, UT, USA). At the 1 kHz frequency, alternating currents spread throughout the extracellular space and impedance increases when extracellular space shrinks,i.e., cells swell (Otsuka et al., 2000). Animals randomly assigned to sham groups underwent surgical procedures with the placement of electrodes without the 2-VO or the induction of CSD.
BrdU injection and immunof l uorescence analysis

Figure 1 Graphic description of ROI placement in brain sections.
Immunohistochemistry was performed to count all newly formed cells in the DG in free fl oating brain sections. Slices were stained with a monoclonal mouse-anti-BrdU (Roche Diagnostics Cooperation; 1:500) and a biotinylated donkey anti-mouse IgG. Fluorescence microscopy was used to calculate the number of double-labeled BrdU- and DCX- or NeuN-positive newly formed cells (neurons) in relation to the total amount of newly formed cells in the DG as previously described (Engelhard et al., 2007).
For immunof l uorescence, free fl oating brain sections were stained with rat-anti-BrdU primary antibody (1:500; rat anti-BrdU, Oxford Biotechnology) and donkey-anti-rat secondary antibody (1:500; Fluorescein Af fi niPure Donkey Anti-Rat IgG, Jackson ImmunoResearch Laboratories, Inc, West Grove, PA, USA). The neuronal markers used were DCX for the 9 days group and NeuN for the 28 days groups). Primary and secondary antibodies for DCX were goat-anti-DCX (1:500; sc-8066, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and donkey-anti-goat (1:500; Rhodamine RedTM-X AffiniPure Donkey Anti-Mouse IgG, Jackson ImmunoResearch Laboratories), respectively. Primary and secondary antibodies for neuron-specif i c nuclear protein or NeuN were mouse-anti-NeuN (1:250; MAB377, Chemicon International, Inc., Billerica, MA, USA) and donkey-anti-mouse (1:500; Rhodamine Red™-X Affini-Pure Donkey Anti-Mouse IgG, Jackson ImmunoResearch Laboratories; see Engelhard et al., 2007). Unlike the DG, the immunof l uorescent-labelled cells were counted in the cortex only within the predef i ned regions of interest (ROI I and II). They were placed in the middle of the cortex between the corpus callosum and the surface of the cortex (Figure 1),i.e., in the area where the 2-VO was induced.e frame dimension of one ROI was set to 220 µm × 160 µm (40× magnif i cations). Two ROIs per section and three sections per hemisphere were analyzed for each animal to assess dif f erences with possible references to migrating cells from “neurogenic” zones towards the cerebral cortex, or perhaps activated resident neural stem cells (NSCs) in the cerebral cortex.e numbers of cells gathered in the examined ROI for respective hemispheres are normalized to mm2.
Statistical analysis
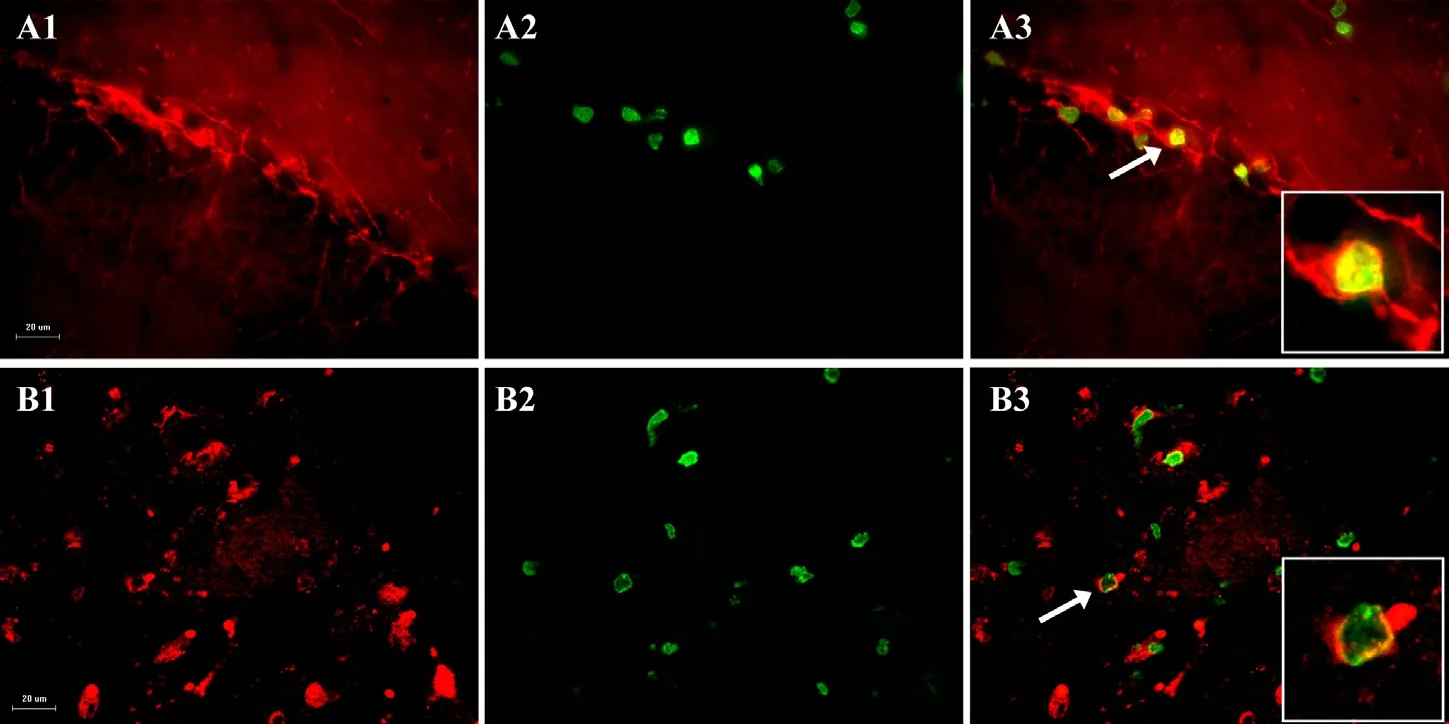
Figure 2 Typical micrographs from dentate gyrus (A) and cortex (B) showing doublecortin-positive cells on day 9 (A1) in the dentate gyrus and neuronal nuclei (NeuN)-positive cells (red) on day 28 (B1) in the parietal cortex of rats with cortical spreading depression plus cerebral venous ischemia.
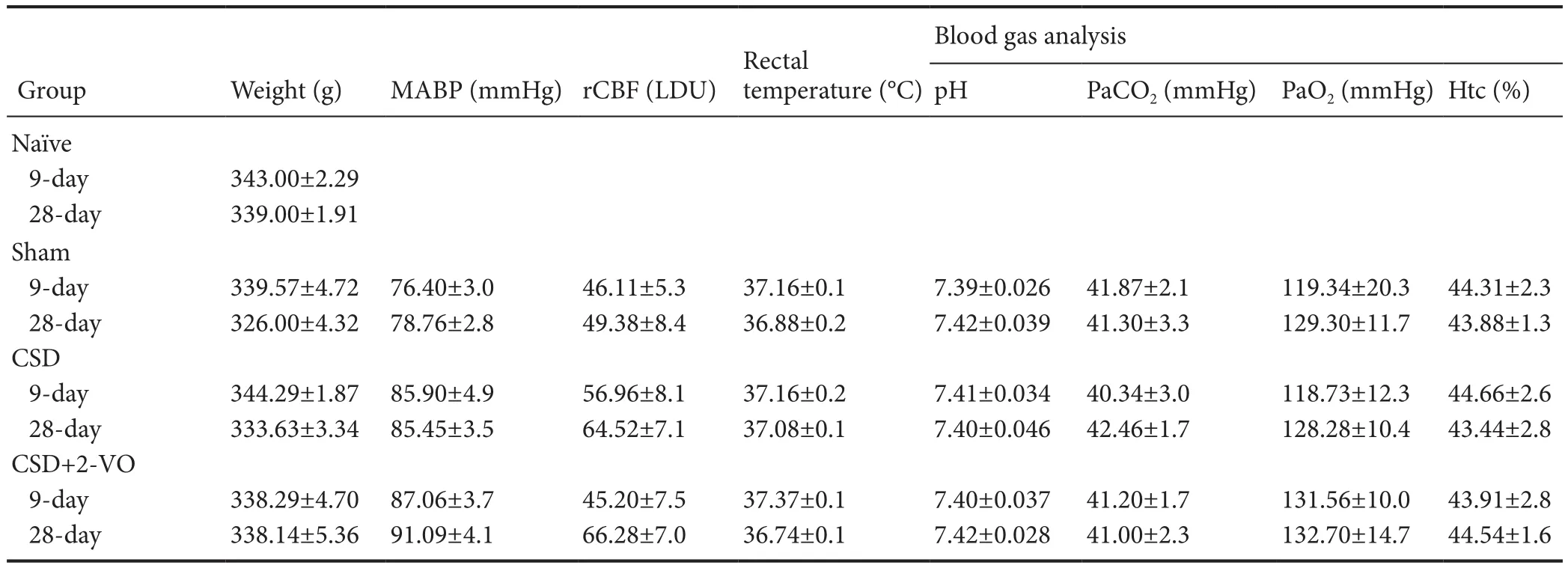
Table 1 Physiological parameters during baseline conditions of rats with CSD + 2-VO
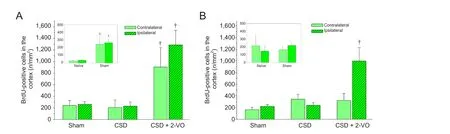
Figure 3 Cell proliferation on days 9 (A) and 28 (B) in the cortex of rats with cortical spreading depression (CSD) plus two-vein oclusion (2-VO).
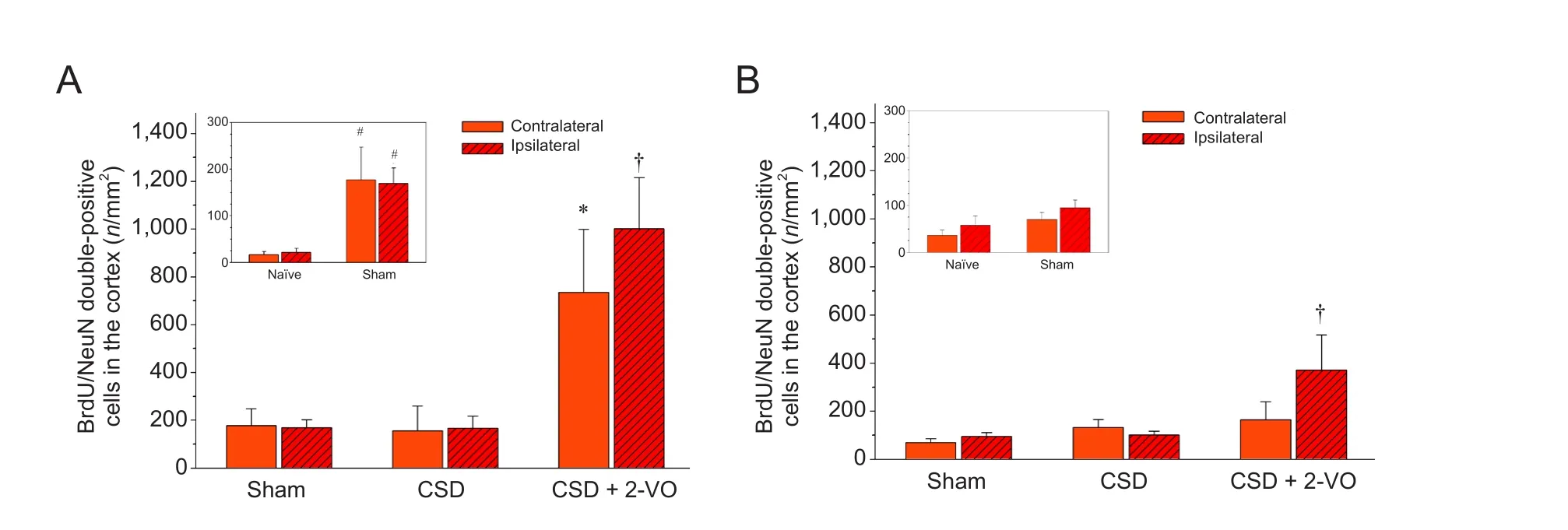
Figure 4 Neurogenesis on days 9 (A) and 28 (B) in the cortex of rats with cortical spreading depression (CSD) plus two-vein occlusion (2-VO).
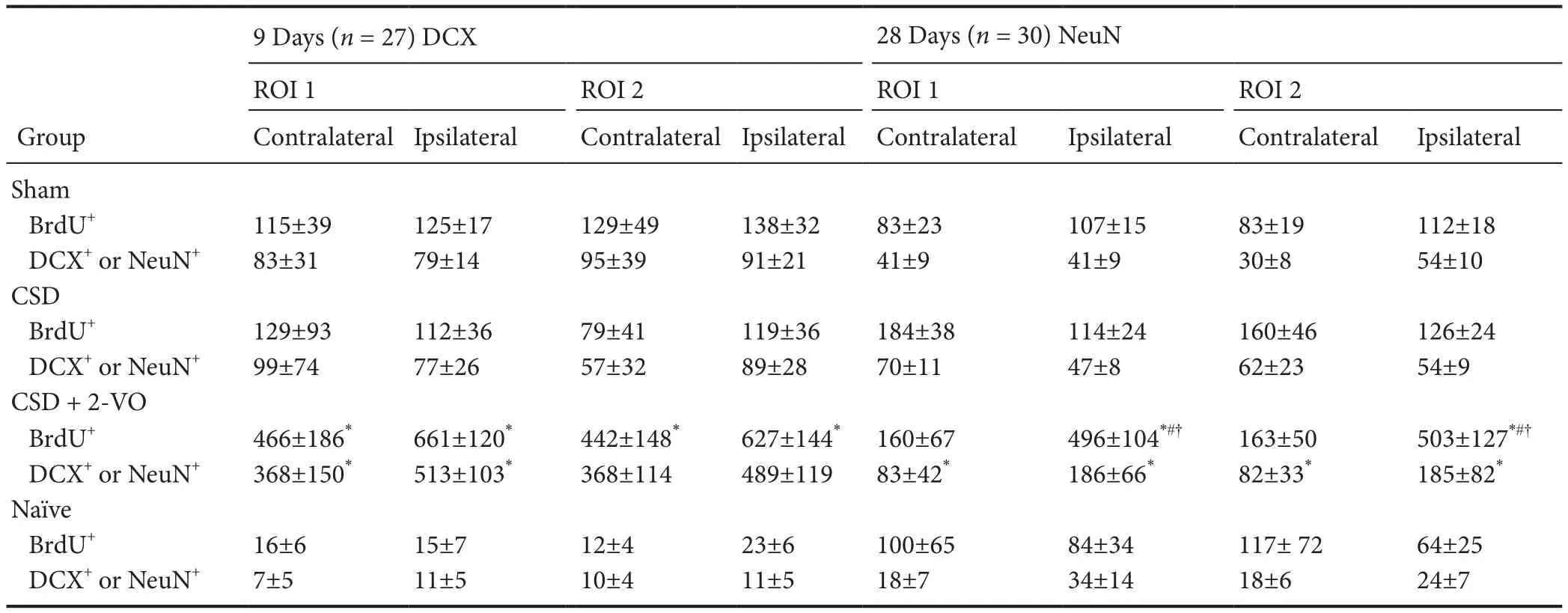
Table 2 Counts per mm2of newly formed BrdU-positive cells and percentages of co-staining with neuronal markers (DCX for 9 days; NeuN for 28 days) in the cerebral cortex of rats subjected to CSD plus 2-VO

Table 3 Counts (n) of newly formed BrdU-positive cells and doublelabeled with neuronal markers DCX for 9 days and NeuN for 28 days in the dentate gyrus
Statistical analysis was performed using SigmaStat®3.5 (Jandel Scientific Software, San Jose, CA, USA). Graphs were done with the graphing soware SigmaPlot Version 10.0 (Systat Software GmbH, Erkrath, Germany). Data and graphs are presented as the mean ± SEM. Sequential changes between sham, CSD, and CSD + 2-VO groups were statistically evaluated using a Kruskal-Wallis H test (one-way analysis of variance on ranks) followed by Dunn’s multiple comparison test for individual group differences. A Student’st-test was performed for comparison between sham-operated and naïve animals. If normality test or equal variance test failed, the Mann-Whitney Rank Sum Test was executed. Statistics were performed on a 5% level of signif i cance.
Results
Physiological data
Immunof l uorescence analysis results
Cerebral cortex
Two def i ned cortical regions of interest (ROI I and ROI II; Figure 1) in neurogenic areas and the expected corticallesion aer 2-VO were determined to investigate changes based on migratory ef f ects.is evaluation did not reveal any dif f erence between the number of newly formed cells or newly formed neurons, neither by spatial nor by temporal pattern (Table 2).us, both ROIs are summarized and presented together. Immunof l uorescence staining (Figure 2) demonstrates an increase of BrdU-positive, newborn cells and double labeled BrdU+/DCX+newborn neurons in both the ipsilateral and contralateral cortex of sham-operated versus naïve animals only at the early observation time (inlet Figures 3A and 4A). Similarly, the combination of CSD + 2-VO led to a signif i cant increase in the numbers of newly formed cells only on day 9 and correspondently, at a lower level, in number of new neurons in the ipsilateral and contralateral cerebral cortex compared to CSD alone or sham operation (Figures 3A and 4A). In contrast, on day 28, cell proliferation and neurogenesis were identified solely in the ipsilateral cortex in the CSD + 2-VO group (Figures 3B and 4B).
DG
Immunohistological analysis in the DG differentiated sham-operated rats from naïve rats by a significantly higher generation of newly formed cells and newly formed neurons in the ipsilateral hemisphere. Moreover, CSD or CSD combined with 2-VO tended to induce more BrdU/DCX double-stabled cells mainly aer 9 days compared to sham operation (Table 3). Nonetheless, there was no statistical difference between sham, CSD and CSD + 2-VO groups. Likewise, the number of newly formed cells and newly formed neurons on day 28 was higher in the ipsilateral DG aer CSD or CSD + 2-VO induction than in naïve or sham operated rats. In the contralateral hemisphere, the number of newly formed cells and neurogenesis were generally more discrete and was increased aer CSD + 2-VO compared to naïve rats (for numbers please see Table 3).
Discussion
Proliferation in the cortex occurred in our study only when CSD was combined with focal venous ischemia. Newly formed cells were increased aer 9 days in the ipsilateral and contralateral hemispheres. Proliferation on day 28 was less pronounced and happened only in the ipsilateral cortex.e similar pattern was detected in neurogenesis showing newly formed neurons aer 9 and 28 days in the parietal cortex in the CSD + 2-VO group only. On day 28, the magnitude was also less pronounced compared to that on day 9. Since CSD alone had no ef f ect, the stimulating trigger for proliferation and formation of newly formed immature neurons has to be induced by ischemic signaling and/or neuronal damage. Furthermore, these fi ndings point out that the ef f ect of CSD depends on the state of the cerebral tissue; in the normal brain, CSD has no pathological consequence to the parenchyma (Nedergaard and Hansen, 1988). Similar to preconditioning when short ischemic episodes before severe injury are able to protect the brain, Yanamoto et al. (2005) have shown that the pretreatment with prolonged KCL-infusion starting 48 hours before focal ischemia in a 3-vessel-occlusion mouse model with partial reperfusion reduced infarction size and had neuroprotective potential. In contrast, an increasing number of spreading depression positively correlates with growing infarct volume in the model of 2-VO (Otsuka et al., 2000) and peri-infarct depolarization increases infarction after middle cerebral artery occlusion (Mies et al., 1993).ese phenomena support the theory that CSD aggravates neuronal damage solely “in tissue at risk”,i.e., in the penumbra, where energy supply is no longer sufficiently sustained to maintain resting metabolism. CSD may trigger proliferation and neurogenesis in a non-neurogenic zone through “inverse” coupling and neuronal damage aer challenging tissue at risk. Likely, Gu et al. (2000) conf i rmed persistent stimulation of neurogenesis in the DG as well as in the “non-neurogenic” cortical layer IV after CSD and photothrombotic stroke. These reports are in line with the literature which suggests spreading depression to be potentially associated with migraines, seizures, head injury and cerebral ischemic infarction (Hadjikhani et al., 2001; Fabricius et al., 2006), ensuring the potential to stimulate persistent neurogenesis or to produce ectopic new neuron-like cells (Yanamoto, 2005; Xue et al., 2009). Although the mechanism is unknown, non-neurogenic regions of the brain such as cortex are proposed to become more permissive to neurogenic processes in pathological conditions, possibly trying to compensate for the loss of neurons.
Interestingly CSD plus ischemia induced proliferation and neurogenesis in the contralateral hemisphere, which disappeared after 28 days. Similar effects have been reported in the contralateral hemisphere aer focal ischemia by middle cerebral artery occlusion (Garbuzova-Davis etal., 2013; Arango-Dávila et al., 2016) proposing different mechanisms for diaschisis.e loss of excitatory af f erent inputs on the corticopontocerebellar pathway may cause contralateral hypoperfusion (Serteser et al., 2001). Remote edema aer middle cerebral artery occlusion is discussed as movement of extravasated protein from the lesion (Izumi et al., 2002). Similar temporal changes with an early cell proliferation up to day 7 which was thereaer followed by a decrease have been reported for the contralateral subventricular zone aer transient middle cerebral artery occlusion (Qi et al., 2007).
There is no consensus whether newly formed neurons originate from stem cells located in “non-neurogenic” cortical layers or migrate from neurogenic zones in the adult brain. The lack of clear differences in the number of cells within our ROI I and II at any evaluated time allows for the following conclusion: Either, the newly formed neurons in the cortex have no migratory background from the neurogenic zones, but are rather derived from cortical regions. Or alternatively, migration does occur from both of this so called neurogenic zones (DG and subventricular zone), but ceases early,i.e., before day 9. In the present study, it is rather unlikely that newly formed neuronal cells originate from the DG because CSD alone or combined with 2-VO compared to sham operated animals did not further raise neurogenesis on days 9 and 28 in the ipsilateral DG but in the cortex.e relative low rate of proliferation and dif f erentiation of cells in the DG seen in our study compared to a solid neurogenesis in the adult DG induced by a continuous infusion of 1µL KCl (4 M) per hour over 48 hours (Urbach et al., 2008) may be explained by the weak signaling from ten induced CSD by 10 mM KCL within a time window of 60 minutes. In addition, the continuous infusion of KCl (4 M, 1 µL/h over 48 hours) induces dividing radial glia, neural progenitor and neural precursor cells in cortical layer I up to day 3 (glial fi brillary acidic protein, vimentin and nestin immunostaining) and new immature neurons (BrdU- and beta-tubulin III; Xue et al., 2009). Although the authors could not identify whether nestin/vimentin positive cells appearing in cortical Layers V to VI are neural precursors originating from cortical layer I, they propose that the astrocytes residing in the subpial zone of the adult cortex are latent or quiescent neural progenitors which can be triggered by multiple SD waves to divide. Yanamoto et al. (2005) detected BrdU-positive and β-tubulin III double-labelled cells in“non-neurogenic regions 48 hours aer SD induced by continuous KCl-infusion.ese cells increased on day 6 and/or day 12 in the caudate putamen, in the frontoparietal cortex (restricted to the cortical layer V–VI) and in the anterior cingulate cortex, but not in the vehicle group. Since these ectopic new neuron-like cells did not express the marker for migrating neuroblasts (at least for 12 days), the authors suggest that these cells are not migrating or dif f erentiating as new neurons from the subventricular zone.
Nonetheless, independent of its origin the determined fate of neuronal progenitors is controlled by signaling molecules within the niche or microenvironment of the brain (Jagasia et al., 2006). Palmer et al. (2000) investigated the proliferation and differentiation of neural stem cells in the hippocampal DG in close proximity of blood vessels, indicating possible participation of endothelial cells in the regulation of neural stem cell proliferation (Shen et al., 2004). Thus, endothelial cells foster conditions leading to local proliferation of endogenous brain cells. Even so, numerous studies suggest that the vast majority of neuroblasts die primarily by apoptotic mechanisms, and only a small population of newly generated neurons in response to ischemic insults survive long enough to mature into potentially functional neurons (Arvidsson et al., 2002; Jin et al., 2003; Zhang et al., 2004).is may explain reductions in the total number of cells aer 28 days as seen in this study. Our study further supports the premise that the combination of CSD and ischemia stabilizes long-term neurogenesis in the cortex. It is clear, however, that for newly arrived cells to survive, the environment must be receptive, provide trophic support and eventually foster their integration within the tissue. Then, synaptic contacts within the surrounding parenchymal circuitry are required for differentiation and survival of newly generated cells (Gould et al., 1999a). Trophic factors like vascular endothelial growth factor, basic fi broblast growth factor and brain-derived neurotrophic factor (BDNF), in addition to being angiogenic, have strong chemotactic ef f ects on stem-like cells and promote the survival and viability of these cells. CSD is known to increase BDNF levels in the brain (Kokaia, 1993; Yanamoto et al., 2005).e amplif i cation of the expression of these trophic factors in neurons and glial cells by spreading depression protects against subsequent focal brain ischemia.is phenomena may contribute to neuroprotection induced by CSD (Matsushima et al., 1998). But our study points out that in combination with neuronal/cortical injury, CSD exacerbates the damage by ischemia. In this case, ischemia starts a wide range of pathways which lead to a microenvironment that favors cell death as well as neurogenesis. Important players aer ischemia are microglia, endothelial cells and blood degradation products. Microglia are activated, move towards the infarct site and secret pro-inf l ammatory cytokines which hinder cell proliferation and neurogenesis in neurogenic regions such as subventricular zone (Solano Fonseca et al., 2016). On the other hand, secretion of anti-inflammatory cytokines such as interleukin-4 by the microglial subpopulation M2 supports proliferation and neurogenesis in a model of transient middle cerebral artery occlusion (Choi et al., 2017). According to a previous study (Kaido et al., 2012), inflammation was induced after CSD + 2-VO, so it could have inf l uenced neurogenesis in this study. Degradation of coagulated blood releases carbon monoxide which has been shown to reduce apoptotic cell death and hence supports neurogenesis (Almeida, 2016).is might also be linked to a necessary contact of stem cells to the laminin protein α6β1 integrin. Ischemia upregulates integrins in the penumbra (Li et al., 2012) and the connection between stem cells and α6β1 integrin has been shown to promote stem cell proliferation and survival in cell culture (Rosa et al., 2016).us, the survival of stem cells aer CSD combined with 2-VO seems tobe a multifactorial mechanism. Further studies are necessary to elucidate the origin of newly formed neurons, pathways which promote neurogenesis and distinguish between ef f ects of 2-VO with CSD and with pharmacologically suppressed CSD.
Results from this study provide evidence for neurogenesis in the cerebral cortex aer CSD and its combination with 2-VO. Our fi ndings have shown that CSD in combination with 2-VO has an early (9 days) stimulating ef f ect on neural stem cells in the cortex, and this effect can be maintained for at least 28 days aer ischemia/CSD.is is likely attributed to the direct (2-VO) and indirect (CSD) ef f ects of tissue ischemia. Our fi ndings suggest that apart from the DG, the microenvironment in the cerebral cortex after CSD + 2-VO supports cell proliferation and differentiation, and also leads to long-term sustainability in the numbers of newly generated cells. We postulate that induced neurogenesis aer CSD especially in the cerebral cortex is rather a direct ef f ect of ischemia rather than CSD in itself.
Acknowledgments:We would like to express our thanks to the stuff members of the Institute for Neurosurgical Pathophysiology: Anett Ehlert (histology), Fatemeh Kafai (secretary), Laszlo Kopacz (electronics) and Michael Malzahn (animal care) for their support.
Author contributions:RT performed animal experiments. SIO was responsible for immunostaining and examination for cell proliferation and neurogenesis. SIO and AH performed data analysis and statistics. BA and OK wrote and revised the paper. All authors approved the fi nal version of this paper.
Conf l icts of interest:None declared.
Research ethics:All animals were treated and used according to the guidelines of the Landesuntersuchungsamt Rheinland-Pfalz (approval No. AZ: 177-07/051-16), which parallel those of the NIH guide for the care and use of animals in scientif i c experimentation.
Data sharing statement:
Plagiarism check:Checked twice by ienticate.
Peer review:Externally peer reviewed.
Open access statement:
Almeida AS, Soares NL, Vieira M, Gramsbergen JB, Vieira HL (2016) Carbon monoxide releasing molecule-A1 (CORM-A1) improves neurogenesis: increase of neuronal dif f erentiation yield by preventing cell death. PLoS One 11:e0154781.
Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O (2002) Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 8:963-970.
Arango-Dávila CA, Muñoz Ospina BE, Castaño DM, Potes L, Umbarila Prieto J (2016) Assessment transcallosal Diaschisis in a model of focal cerebral ischemia in rats. Colomb Med (Cali) 47:87-93.
Bernier PJ, Bedard A, Vinet J, Levesque M, Parent A (2002) Newly generated neurons in the amygdala and adjoining cortex of adult primates. Proc Natl Acad Sci U S A 99:11464-11469.
Carleton A, Petreanu LT, Lansford R, Alvarez-Buylla A, Lledo PM (2003) Becoming a new neuron in the adult olfactory bulb. Nat Neurosci 6:507-518.
Choi JY, Kim JY, Kim JY, Park J, Lee WT, Lee JE (2017 ) Phenotype Microglia-derived cytokine stimulates proliferation and neuronal dif f erentiation of endogenous stem cells in ischemic brain. Exp Neurobiol 26:33-41.
Cooper-Kuhn CM, Kuhn HG (2002) Is it all DNA repair? Methodological considerations for detecting neurogenesis in the adult brain. Brain Res Dev Brain Res 134(1-2):13-21.
Engelhard K, Winkelheide U, Werner C, Kluge J, Eberspächer E, Hollweck R, Hutzler P, Winkler J, Kochs E (2007) Sevof l urane af f ects neurogenesis aer forebrain ischemia in rats. Anesth Analg 104:898-903.
Fabricius M, Fuhr S, Bhatia R, Boutelle M, Hashemi P, Strong AJ, Lauritzen M (2006) Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain 129:778-790.
Garbuzova-Davis S, Rodrigues MC, Hernandez-Ontiveros DG, Tajiri N, Frisina-Deyo A, Bof f eli SM, Abraham JV, Pabon M, Wagner A, Ishikawa H, Shinozuka K, Haller E, Sanberg PR, Kaneko Y, Borlongan CV (2013) Blood-brain barrier alterations provide evidence of subacute diaschisis in an ischemic stroke rat model. PLoS One. 8:e63553.
Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ (1999a) Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci 2:260-265.
Gould E, Reeves AJ, Graziano MS, Gross CG (1999b) Neurogenesis in the neocortex of adult primates. Science 286:548-552.
Gu W, Brannstrom T, Wester P (2000) Cortical neurogenesis in adult rats aer reversible photothrombotic stroke. J Cereb Blood Flow Metab 20:1166-1173.
Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA (2001) Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A 98:4687-4692.
Heimann A, Takeshima T, Alessandri B, Noppens R, Kempski O (2003) Effect of hypertonic/hyperoncotic treatment after rat cortical vein occlusion. Crit Care Med 31:2495-2501.
Izumi Y, Haida M, Hata T, Isozumi K, Kurita D, Shinohara Y (2002) Distribution of brain oedema in the contralateral hemisphere aer cerebral infarction: repeated MRI measurement in the rat. J Clin Neurosci 9:289-293.
Jagasia R, Song H, Gage FH, Lie DC (2006) New regulators in adult neurogenesis and their potential role for repair. Trends Mol Med 12:400-405.
Jin K, Sun Y, Xie L, Peel A, Mao XO, Batteur S, Greenberg DA (2003) Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol Cell Neurosci 24:171-189.
Kaido T, Kempski O, Heimann A, Heers C, Bartsch D (2012) Cluster analysis of mRNA expression levels identif i es multiple sequential patterns following focal cerebral ischemia. Turk Neurosurg 22:441-447.
Kaplan MS (1981) Neurogenesis in the 3-month-old rat visual cortex. J Comp Neurol 195:323-338.
Kempski O, Seiwert T, Otsuka H, Heimann A, Nakase H (1999) Modelling of the ischemic penumbra. Acta Neurochir Suppl 73:41-44.
Kokaia Z, Gidö G, Ringstedt T, Bengzon J, Kokaia M, Siesjö BK, Persson H, Lindvall O, (1993) Rapid increase of BDNF mRNA levels in cortical neurons following spreading depression: regulation by glutamatergic mechanisms independent of seizure activity. Brain Res Mol Brain Res 19:277-286.
Leão AA (1944) Spreading depression of activity in the cerebral cortex. J Neurophysiol 7:359-390.
Li L, Liu F, Welser-Alves JV, McCullough LD, Milner R (2012) Upregulation of fi bronectin and the α5β1 and αvβ3 integrins on blood vessels within the cerebral ischemic penumbra. Exp Neurol 233:283-291.
Lois C, Alvarez-Buylla A (1994) Long-distance neuronal migration in the adult mammalian brain. Science 264:1145-1148.
Luzzati F, De Marchis S, Fasolo A, Peretto P (2007) Adult neurogenesis and local neuronal progenitors in the striatum. Neurodegener Dis 4:322-327.
Matsushima K, Schmidt-Kastner R, Hogan MJ, Hakim AM (1998) Cortical spreading depression activates trophic factor expression in neurons and astrocytes and protects against subsequent focal brain ischemia. Brain Res 807:47-60.
Mies G, Iijima T, Hossmann KA (1993) Correlation between peri-infarct DC shis and ischaemic neuronal damage in rat. NeuroReport 4:709-711.
Nacher J, Crespo C, McEwen BS (2001) Doublecortin expression in the adult rat telencephalon. Eur J Neurosci 14:629-644.
Nakase H, Heimann A, Kempski O (1996) Local cerebral blood flow in a rat cortical vein occlusion model. J Cereb Blood Flow Metab 16:720-728.
Nakase H, Kempski O, Heimann A, Takeshima T, Tintera J (1997) Microcirculation aer cerebral venous occlusions as assessed by laser Doppler scanning. J Neurosurg 87:307-314.
Nedergaard M, Hansen AJ (1988) Spreading depression is not associated with neuronal injury in the normal brain. Brain Res 449:395-398.
Otsuka H, Ueda K, Heimann A, Kempski O (2000) Ef f ects of cortical spreading depression on cortical blood flow, impedance, DC potential, and infarct size in a rat venous infarct model. Exp Neurol 162:201-214.
Palmer TD, Willhoite AR, Gage FH (2000) Vascular niche for adult hippocampal neurogenesis. J Comp Physiol 425:479-494.
Qi CF, Zhang JS, Tian YM, Chen XL, Zhang PB, Xiao XL, Qiu F, Liu Y (2007) Effect of ligustrazine on cell proliferation in subventricular zone in rat brain with focal cerebral ischemia-reperfusion injury. J Cent South Univ (Med Sci) 32:396-400.
Rosa AI, Grade S, Santos SD, Bernardino L, Chen TC, Relvas J, Hofman FM, Agasse F (2016) Heterocellular contacts with mouse brain endothelial cells via laminin and α6β1integrin sustain subventricular zone (SVZ) stem/progenitor cells properties. Front Cell Neurosci 10:284.
Serteser M, Ozben T, Gümüşlü S, Balkan S, Balkan E (2001) Biochemical evidence of crossed cerebellar diaschisis in terms of nitric oxide indicators and lipid peroxidation products in rats during focal cerebral ischemia. Acta Neurol Scand 103:43-48.
Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S (2004) Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 304:1338-1340.
Solano Fonseca R, Mahesula S, Apple DM, Raghunathan R, Dugan A, Cardona A, O’Connor J, Kokovay E (2016) Neurogenic niche microglia undergo positional remodeling and progressive activation contributing to age-associated reductions in neurogenesis. Stem Cells Dev 25:542-55.
Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M (2007) Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci 10:754-762.
Tamura Y, Kataoka Y, Cui Y, Yamada H (2004) Cellular proliferation in the cerebral cortex following neural excitation in rats. Neurosci Res 50:129-133.
Urbach A, Redecker C, Witte OW (2008) Induction of neurogenesis in the adult dentate gyrus by cortical spreading depression. Stroke 39:3064-3072.
Xue JH, Yanamoto H, Nakajo Y, Tohnai N, Nakano Y, Hori T, Iihara K, Miyamoto S (2009) Induced spreading depression evokes cell division of astrocytes in the subpial zone, generating neural precursor-like cells and new immature neurons in the adult cerebral cortex. Stroke 40:e606-613.
Yanamoto H, Miyamoto S, Tohnai N, Nagata I, Xue JH, Nakano Y, Nakajo Y, Kikuchi H (2005) Induced spreading depression activates persistent neurogenesis in the subventricular zone, generating cells with markers for divided and early committed neurons in the caudate putamen and cortex. Stroke 36:1544-1550.
Zhang R, Zhang Z, Zhang C, Zhang L, Robin A, Wang Y, Lu M, Chopp M (2004) Stroke transiently increases subventricular zone cell division from asymmetric to symmetric and increases neuronal dif f erentiation in the adult rat. J Neurosci 24:5810-5815.
Edited by Li CH, Song LP, Zhao M
*< class="emphasis_italic">Correspondence to: Axel Heimann, Doctor of Veterinary Medicine (DVM), axel.heimann@ unimedizin-mainz.de.
Axel Heimann, Doctor of Veterinary Medicine (DVM), axel.heimann@ unimedizin-mainz.de.
#
orcid: 0000-0003-2510-3228 (Heimann Axel)
10.4103/1673-5374.213547
Accepted: 2017-07-08
no intervention except 5-bromo-2′-deoxyuridine (BrdU) treatment for 7 days. Sagittal brain slices (40 µm thick) were co-stained for BrdU and doublecortin (DCX; new immature neuronal cells) on day 9 or NeuN (new mature neuronal cells) on day 28. On day 9 aer sham operation, cell proliferation and neurogenesis occurred in the cortex in rats.e sole induction of CSD had no ef f ect. But on days 9 and 28, more proliferating cells and newly formed neurons in the ipsilateral cortex were observed in rats subjected to CSD + 2VO than in rats subjected to sham operation. On days 9 and 28, cell proliferation and neurogenesis in the ipsilateral dentate gyrus was increased in sham-operated rats than in naïve rats. Our data supports the hypothesis that induced cortical neurogenesis aer CSD + 2-VO is a direct ef f ect of ischemia, rather than of CSD alone.
- 中国神经再生研究(英文版)的其它文章
- Transcriptional inhibition in Schwann cell development and nerve regeneration
- A progressive compression model of thoracic spinal cord injury in mice: function assessment and pathological changes in spinal cord
- Effects of estrogen receptor modulators on cytoskeletal proteins in the central nervous system
- Optogenetics and its application in neural degeneration and regeneration
- Live-cell imaging: new avenues to investigate retinal regeneration
- Neurotrophic factors and corneal nerve regeneration