
乳腺癌癌症干细胞的特异基因识别
2017-08-16 10:26郭鹏飞贺平安
浙江理工大学学报(自然科学版) 2017年2期
郭鹏飞,贺平安
(浙江理工大学理学院,杭州 310018)
乳腺癌癌症干细胞的特异基因识别
郭鹏飞,贺平安
(浙江理工大学理学院,杭州 310018)
乳腺癌是一种严重威胁女性健康的恶性肿瘤,癌症干细胞假说的提出为乳腺癌的起因以及治疗提供了新的模型。对746个乳腺癌样本中的18409个基因和1035个miRNAs,通过生物信息学方法构建共表达网络,将其划分到不同的共表达模块中;利用胚胎干细胞和间充质干细胞的特性进一步筛选模块,得到两个大小分别为2019和859且与上述两类干细胞相关的基因集;最后通过构建这两个基因集的调控网络,筛选出两个胚胎干细胞的特异性关键基因TPX2和MCM10,以及间充质干细胞的特异基因COL5A2。这些基因可以作为癌症干细胞的候选特异性标志物,有望成为潜在的乳腺癌治疗标靶。
乳腺癌;胚胎干细胞;间充质干细胞;基因调控网络;关键基因
0 引 言
乳腺癌是全世界女性最常见的一种恶性肿瘤,其发病率占女性全身其他恶性肿瘤的10%左右。据统计,每年约有120万新发乳腺癌病例,乳腺癌已成为女性发病率最高的癌症[1]。随着医学的发展和治疗手段的进步,乳腺癌患者的生存率已经得到很大改善,但它的耐药性以及预后复发等问题依然困扰着许多研究者。
癌症干细胞假说认为癌症是由一小群癌症干细胞造成的,即癌症干细胞是癌症异常增殖、侵袭、转移、耐药以及复发等的根源。Wicha等[2]利用不同的细胞表面标记物对细胞进行标记,验证了乳腺癌中存在着癌症干细胞,这为癌症干细胞的研究提供了坚实基础。此后,Takebe等[3]研究发现Notch、Hedgehog (HH) 和Wnt等细胞通路在癌症干细胞的致癌方面起着至关重要的作用,并对通过抑制这些通路从而控制干细胞复制、存活和分化这一新治疗策略进行了研究。
此外,许多研究者从基因层面研究癌症干细胞涉及到的相关生物过程。Xu等[4]研究发现miR-214在调控卵巢癌干细胞性质方面起着至关重要的作用,并且miR-214可作为治疗卵巢癌的潜在的治疗靶标。Li等[5]研究发现长链非编码RNA(long noncoding RNAs, lncRNAs)能够抑制其标靶mRNA在胶质母细胞瘤干细胞(glioblastoma stem cells, GSCs)中分化,并利用这种方法识别出一些可用来治愈GSCs的候补lncRNAs。此外,Kalamohan等[6]应用加权的基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)和富集度分析的方法分析胃癌的mRNA数据,发现胃癌的两种亚型分别与不同的干细胞特征相关。
以上研究表明,癌症干细胞假说能更好地解释癌症的起源,并指导癌症治疗。本文基于以上研究的成果,利用多种生物信息学方法分析现有数据库中乳腺癌的基因表达谱数据,期望可以辨别出某些与癌症干细胞相关的关键基因。该研究结果有助于人们更好地理解乳腺癌的发生、发展机制。
1 数据和方法
1.1 数据及数据预处理
本文研究的乳腺癌的miRNASeq、RNASeqV2数据以及乳腺癌患者的临床数据,均下载于The Cancer Genome Atlas (TCGA)数据库,其中RNASeqV2和miRNASeq数据是分别通过RNA测序和miRNA测序得到的样本的基因表达和miRNA表达数据。
由于某些miRNAs可以通过调控基因表达进而参与调控细胞分化和癌症生成等重要生命过程,故本文合并两种数据用于研究分析。首先,如果某个基因或miRNA在超过50%的样本中其原始数据缺失,则将该基因或miRNA删除;然后使用LIMMA package[7]将处理过的基因和miRNAs原始数据转化为基因表达值,并将二者合并;最后使用反分位数归一化(inversely normalize)的方法处理合并的基因表达数据,使其处于同一水平进行后续分析。经过预处理后得到一个由746个乳腺癌样本中的18409个基因和1035个miRNAs的表达值构成的数据集D=(dij)19444×746,其中dij表示第i个基因在第j个样本中的表达值。
另外,从The Gene Expression Omnibus (GEO)数据库下载了胚胎干细胞(GSE29625)和间充质干细胞(GSE28974)的mRNA表达谱数据。
1.2 构建基因共表达网络
WGCNA算法[8]是一种从表达谱数据中挖掘模块(module)信息的算法。在该算法中模块被定义为一组具有类似表达谱的基因,即如果某些基因在一个生理过程或不同组织中总是具有相类似的表达变化,则将其定义为一个模块。

(1)
(2)
(3)

另外考虑到基因i可以通过基因μ与基因j相互作用,故将邻接矩阵被转换成拓扑矩阵Ω=(wij),其中:
(4)
这里lij=∑uaiuauj,表示与基因i、j都相邻的基因μ之间的邻接系数乘积和;ki=∑uaiu为基因i单独连接的节点的邻接系数的和。
理论上,胚胎干细胞和间充质干细胞在原发性肿瘤,以及癌细胞的循环过程和转移器官中都可以检测到。因此本文选择这两种干细胞为代表通过基因集的富集度分析[9],查找与癌症干细胞相关的基因。为此,从MSigDB(molecular signatures database)数据库[10]下载了15个与胚胎干细胞特性相关的基因集以及10个跟间充质干细胞特征相关的基因集,作为背景基因进行富集度分析。
基因集的富集度分析基于超几何分布,服从超几何分布(k-1,K,N-K,n)的概率p可通过式(5)来计算:
(5)
其中:n表示模块中基因的个数;K表示与胚胎干细胞特性相关的基因集或者跟间充质干细胞特征相关的基因集中基因的个数;k和N分别为上述模块和基因集的交集和并集中基因的个数。错误发现率[11](false discovery rate,FDR)用于评价基因模块是否富集于与两种干细胞相关的基因集。
1.4 优化与癌症干细胞相关的基因集
通过基因集的富集度分析得到两个分别与胚胎干细胞和间充质干细胞相关的基因集,为了优化这两个基因集,本文应用多尺度嵌合基因共表达网络分析(multiscale embedded gene co-expression network analysis, MEGENA)算法[12]对二者重新进行精确分类。
MEGENA算法首先计算任意两个基因之间的相关性,并依据相关性的大小对基因对排序;接着通过平面最大过滤图算法(planar maximally filtered graph , PMFG)将其嵌入拓扑网络,从而构建平面滤波网络(planar filtered networks, PFNs);然后通过最短路径距离、本地路径索引和整体模块性三个标准对最初的PFNs进行多次迭代处理,得到更精确的分类。
1.5 基因表达的调控网络的建立
随机森林算法[13]是一种基于决策树模型的算法,它主要通过一个重要性评分矩阵来推断调控网络。相比于其他构建调控网络的方法,随机森林算法可以得到一个有向的调控网络,使得基因间的调控关系更加明确。故本文利用随机森林算法在与癌症干细胞相关的基因集中构建基因调控网络。
随机森林算法将预测n个基因间的调控网络的问题转化为求解n个不同的回归问题。首先选取一个基因作为靶基因(因变量),其余n-1个基因作为输入基因(自变量),做回归分析预测靶基因。每个输入基因在预测靶基因过程中计算相应的变量重要性评分(variable importance measure,VIM),并以此作为推定基因间调控关系的指标。将得到的所有靶基因与输入基因之间的调控关系依据其大小排序,从而构造调控网络。本文用R语言中的randomForest package[14]构建有向的基因调控网络,同时用Cytoscape软件[15]实现基因调控网络的可视化。
在有向的基因调控网络中,基因的顶点出度是以该点为起点的边的个数。本文根据基因的顶点出度的大小筛选与癌症干细胞相关的关键基因。
1.6Kaplan-Meier生存分析
生存分析是将事件的结果和出现这一结果所经历的时间结合起来分析的一种统计分析方法[16]。Kaplan-Meier生存分析将乘积极限法应用于临床数据中样本生存或死亡这两种状态所对应的生存时间,从而计算出样本的生存率及其标准误差。然后利用log-rank检验来比较两组或多组生存率,并通过p-value来评价不同组的生存率是否相同。
对得到的关键基因,本文通过构建Kaplan-Meier生存曲线来验证它们对乳腺癌的重要性。
2 结 果
2.1 表达数据的聚类分析
利用WGCNA算法,输入数据集D中的数据,首先计算任意两个基因之间的皮尔森相关性系数得到相关性矩阵。接着通过无尺度网络原则确定β。如图1所示,当β=5时R2= 0.8220,因此本文选择β=5作为加权系数将相关性矩阵转化为邻接矩阵。最后利用节点的相异程度进行分层聚类,结果746个乳腺癌样本中的18409个基因和1035个miRNAs被聚类到47个不同的模块中。47个模块分别被记作M1—M47,并且每个模块的大小从33到2598数目不等。由于这些共表达基因倾向于功能相关的,故这种聚类方式也意味着乳腺癌的转录组包括47个不同或相关的生物过程。而这些生物过程有助于研究乳腺癌中的分子机制和关键性驱动因子,因此这些模块值得深入研究[6]。

图1 加权系数 β的选取
2.2 识别与胚胎干细胞和间充质干细胞相关的模块
将WGCNA算法得到的模块和MSigDB数据库中下载的与两种干细胞特性相关的基因集作为输入数据进行富集度分析,本文以FDR<0.05为标准来确定结果。
在与胚胎干细胞特性相关的基因集的富集度分析结果中,有8个基因模块与胚胎干细胞特性相关,它们分别是M1—M8,具体结果见表1。另一方面,有6个模块富集于间充质干细胞,分别为M2、M5、M6、M8、M9和M10,其结果如表2所示。

表1 与胚胎干细胞性质相关的模块的富集度分析结果

表2 与间充质干细胞特征相关的模块的富集度分析结果
在表1和表2中,第一列和第二列分别表示模块及其大小,第四列是每个模块与从MSigDB数据库中得到的任一基因集进行一次富集度分析得到的FDR值,第三列是根据FDR的值从小到大的排序,第五列是每个基因模块中富集于MSigDB数据库中的基因集。
为了进一步分析这些基因,本文将与胚胎干细胞性质相关的8个模块合并成一个基因集E1,内含5751个基因;同时将富集于间充质干细胞的6个模块合并成一个基因集E2,内含3441个基因。
2.3 分别确定与胚胎干细胞和间充质干细胞相关的基因集
在WGCNA算法的结果中,有4个模块M2、M5、M6、M8同时富集于胚胎干细胞和间充质干细胞,这使得基因集E1和E2中包含许多相同的基因。为了优化上述两个基因集,本文对这些基因模块作如下处理:
首先,合并两个基因集E1和E2得到新的基因集E3,它包含6102个基因。对于E3中的基因,根据由乳腺癌样本中基因和miRNAs的表达值构成的数据集D构造它的一个子矩阵D1=(dij)6102×746。使用MEGENA算法对该数据重新分类得到新的模块,并对新的模块进行基因集富集度分析,重新筛选出与两种干细胞相关的模块,最后合并模块得到新的与两种干细胞相关的基因集。在这一过程中,本文得到两个基因个数分别为2009和572的且与胚胎干细胞和间充质干细胞相关的基因集F1和F2。
其次,对基因集E1中的基因,重复上述过程,得到分别由1824和802个基因构成的与胚胎干细胞和间充质干细胞相关的基因集F3和F4,并且F3和F4无交集。
然后,对基因集E2中的基因,重复上述过程,得到两个分别与胚胎干细胞和间充质干细胞相关的无交集基因集F5和F6,其大小分别为57和386。
最后,取F1、F3和F5的并集,得到一个包含2019个与胚胎干细胞相关的基因集G1;取F2、F4和F6的并集,得到一个大小为859的与间充质干细胞相关的基因集G2。而且新得到的基因集G1和G2没有交集。表3—表4为上述三组数据利用MEGENA算法分类后,新的模块进行富集度分析的结果。

表3 MEGENA算法分类与胚胎干细胞性质相关的模块的富集度分析结果

表4 MEGENA算法分类与间充质干细胞特征相关的模块的富集度分析结果
2.4 验证基因集G1和G2
为了进一步验证上述过程得到的两个与胚胎干细胞和间充质干细胞相关的基因集。首先合并从GEO数据库下载的癌症胚胎干细胞(GSE29625)和间充质干细胞(GSE28974)的mRNA表达谱数据,然后分别使用分位数归一化[17]和反分位数归一化的方法处理合并后的数据,最终得到一个由10195个基因构成的基因表达数据T1=(tij)10195×24,这里tij为第i个基因在第j个样本中的表达值。T1用于验证上述过程得到的两个与胚胎干细胞和间充质干细胞相关的基因集G1和G2。
图2(a)是由与胚胎干细胞相关的基因集G1在数据T1中的表达值构成的热图,图中每个小方格表示一个基因在样本中的表达量,颜色表示表达量的大小。其中首字母为E的是GSE29625中的样本,首字母为M的是GSE28974中的样本。图中结果表明该基因集的大部分基因在整合数据中的GSE29625样本中具有显著的高表达,在GSE28974样本中具有显著的低表达。类似地,图2(b)是由与间充质干细胞相关的基因集在整合数据T1中的表达值构成的热图。该基因集中的大部分基因在整合数据中的GSE28974样本中具有显著的高表达。


图2 与胚胎干细胞相关的基因集和与间充质干细胞的基因集在两种干细胞的整合数据中的表达模式
基因集G1和G2在整合数据T1中的不同表达模式表明二者与两类干细胞具有明显的相关性,进一步说明使用本文方法得到的结果具有较强的可靠性。
2.5 构建基因调控网络
对与胚胎干细胞相关的基因集G1中的2019个基因,利用其在数据集D中的表达值,使用R语言中的randomForest package[14]构建它们的调控网络。随机森林算法中最重要的参数有两个:一个是建立决策树的个数,本文取1000;另一个是每个节点可选择的候选输入基因个数,在本文中该参数取全部输入基因数的平方根。
利用多重假设检验,求出重要性评分矩阵的每一个值的FDR值,首先取FDR<0.01的边来控制调控网络的大小,得到一个包含53298条边的调控网络。图3是由此网络中全部基因之间的VIM值绘制的直方图,从图中可以看出大部分基因间的重要性评分值小于0.01。为了进一步控制调控网络的规模,本文仅选取基因之间的重要性评分值大于0.01的边。最后得到了一个含有15720条边的有向调控网络。

图3 53298个基因对的 VIM值的频率分布直方图
在构造的有向调控网络中,如果一个基因同时调控多个基因,那么它肯定在某个生物过程中起重要作用,所以文本重点关注那些处于调控关系上游的基因。本文使用R语言中的igraph package[18]计算网络中每个顶点的出度,并根据其大小进行排序。在这个有向调控网络中,TPX2、MCM10、CEP55、BUB1、NCAPG、NCAPH和BUB1B等基因有较大的顶点出度,具体结果如表5所示。特别是TPX2和MCM10这两个基因调控下游基因的个数都超过110,所以本文认为这两个基因是与胚胎干细胞相关的关键基因。图4(a)—(b)是以基因TPX2和MCM10为核心,以及它们调控的基因之间的调控关系构成的调控子网络。为了显示清晰,该调控子网络中仅画出了VIM值大于0.02的边。

表5 基因集G1调控网络中Top12基因的顶点出度
对于包含859个与间充质干细胞相关的基因集G2,本文重复同样的过程。结果发现,在与间充质干细胞相关的基因集的调控网络中,COL5A2、FBN1和COL1A2等基因具有较大的顶点出度,具体结果如表6所示。其中COL5A2基因调控的基因数超过100。因此,基因COL5A2被看作是与间充质干细胞相关的关键基因。它的调控子网络见图4(c)。

(a)TPX2
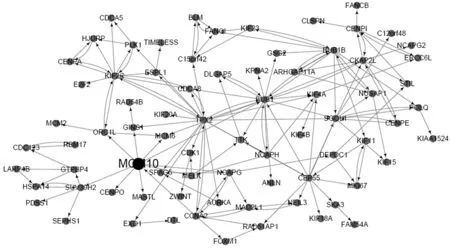
(b)MCM10

(c)COL5A2图4 三个关键基因及其下游调控基因构成的调控子网络

基因顶点出度基因顶点出度COL5A2102COL6A371FBN187VCAN67COL1A287LUM67COL3A174BNC266THBS272CDH1165
由于上述3个基因在相应的调控网络中具有极高的顶点出度,说明TPX2、MCM10和COL5A2在癌症的胚胎干细胞和间充质干细胞的自我更新、分化过程中具有重要作用。故上述三个基因可作为辨别乳腺癌的癌症干细胞的特征基因,以及治愈乳腺癌的潜在的生物靶基因。
2.6 关键基因的生物学分析
本文将3个关键基因的表达谱数据与TCGA数据库中的临床数据整合成一个由657个样本构成的新数据T2进行Kaplan-Meier生存分析,从而进一步研究关键基因的表达方式对乳腺癌患者的生存率的影响,结果如图5所示。在图5中,对应的曲线分别为在基因TPX2和MCM10的高表达和低表达情况下乳腺癌患者的生存曲线,其中x轴表示乳腺癌患者的生存时间,y轴表示患者的生存率;event=1代表患者死亡。观察图5发现处于TPX2和MCM10高表达组的癌症患者相比于低表达组的患者有明显高的死亡率。而且假设检验的p-value都小于0.05,也表明关键基因的不同表达方式对乳腺癌患者生存率的影响显著不同。
基因COL5A2没有上述结论,但Weng等[19]通过研究血小板反应蛋白2(thrombospondin2,THBS2)的表达模式在肺癌发展中的作用,发现COL5A2基因作为THBS2的一个共表达基因,它们的高表达使得肺癌患者具有较低存活率。此外,Fischer等[20]通过对比胶原蛋白的基因在结肠直肠癌患者和正常结肠上皮的组织样品中的差异表达,发现基因COL5A2在基质中的表达与结肠直肠癌相关。Zhang等[21]利用TCGA数据库中的卵巢癌数据构建了贝叶斯网络,其中基因COL5A2同样被发现是关键基因。
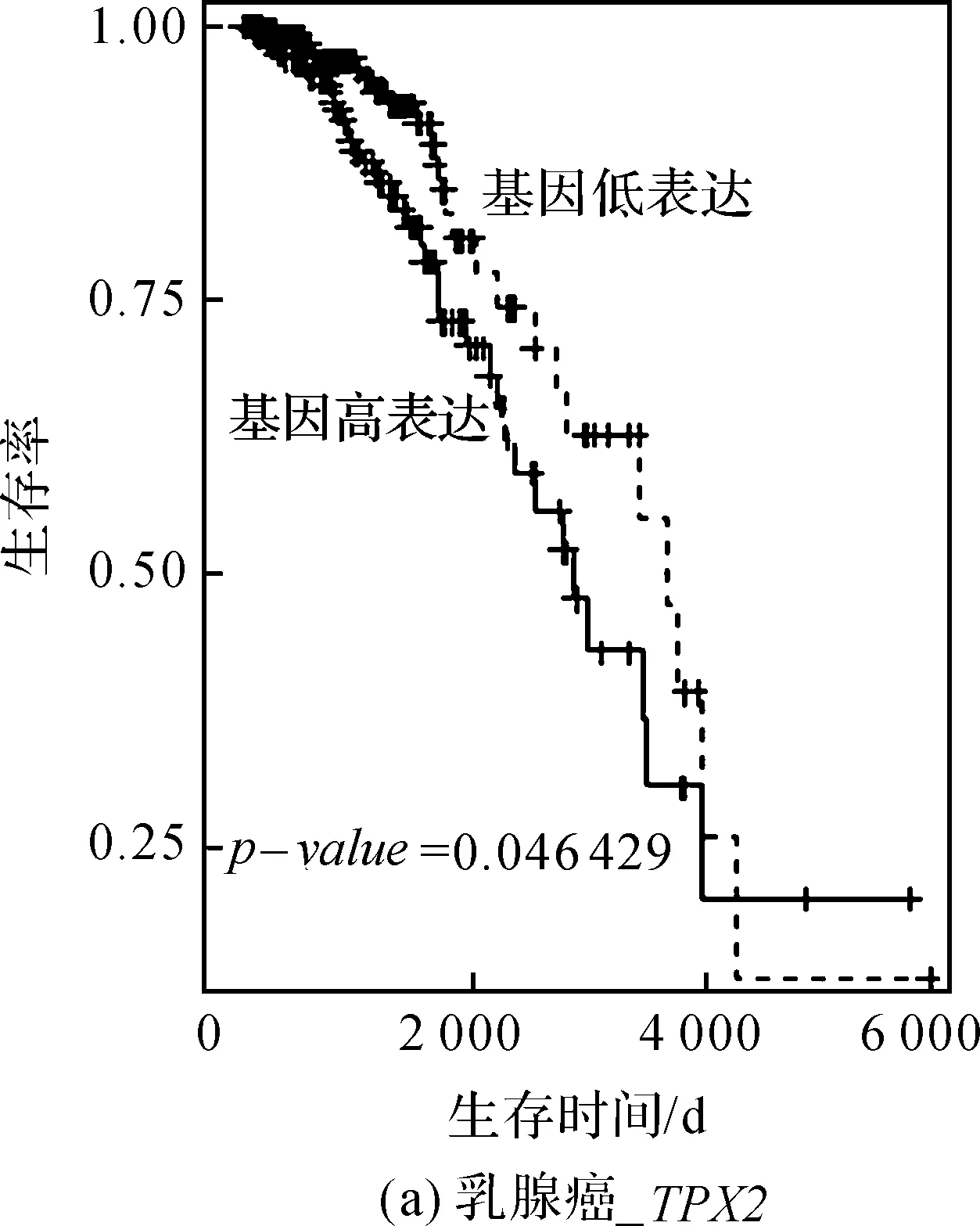

图5 关键基因在乳腺癌临床数据中当event=1时的生存分析
3 结 论
癌症干细胞假说认为癌症很可能起源于干细胞的非正常分化,那么通过中断癌症干细胞的自我更新从而造成其自我更新障碍,完全可以成为一种治疗癌症的理想方式。本文基于这一思想,利用生物信息学中WGCNA算法和MEGENA算法分析乳腺癌的基因和miRNA混合表达数据,将其划分为具有不同或相似生物功能的基因共表达模块,同时利用MSigDB数据库中与胚胎干细胞特性和间充质干细胞特征相关的基因集进行富集度分析,得到了两个分别与乳腺癌胚胎干细胞和间充质干细胞相关的且由2019个基因和859个基因组成的基因集。另外从GEO数据库中下载了癌症胚胎干细胞和间充质干细胞的mRNA表达谱数据,并通过查看上述两个基因集在mRNA表达谱数据中的表达模式,验证了本文得到的两个基因集是可靠的。
进一步利用随机森林算法在上述两个基因集中构建有向的调控网络。通过调控网路的分析,发现了3个在癌症的胚胎干细胞和间充质干细胞自我更新、分化过程中起重要作用的关键基因:TPX2、MCM10和COL5A2。生存分析和已有的结果进一步说明这三个基因是与乳腺癌密切相关的,可以作为治疗乳腺癌的潜在的治疗靶点。
[1] BENSON J R, JATOI I, KEISEH M, et al. Early breast cancer [J]. Lancet,2009,373(9673):1463-1479
[2] WICHA M S, DONTU G, AL-HAJJ M, et al. Stem cells in normal breast development and breast cancer[J].Breast Cancer Research,2003,36(1):59-72.
[3] TAKEBE N, MIELE L, HARRIS P J, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update [J].Nature Reviews Clinical Oncology,2015,12(8):445-464.
[4] XU C X, XU M, TAN L, et al. MicroRNA miR-214 regulates ovarian cancer cell stemness by targeting p53/Nanog [J].Journal of Biological Chemistry,2012,287(42):34970-34978.
[5] LI H. Differential long non-coding RNA and mRNA expression in differentiated human glioblastoma stem cells [J].Mol Med Rep,2016,14(3):2067-2076.
[6] KALAMOHAN K, PERIASAMY J, BHASKAR R D, et al. Transcriptional co-expression network reveals the involvement of varying stem cell features with different dysregulations in different gastric cancer subtypes [J].Molecular Oncology,2014,8(7):1306-1325.
[7] LAW C W, CHEN Y, SHI W, et al. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts [J].Genome Biology,2014,15(2):1417.
[8] 宋长新,雷萍,王婷.基于WGCNA算法的基因共表达网络构建理论及其R软件实现[J].基因组学与应用生物学,2013,32(1):135-141.
[9] RIVALS I, PERSONNAZ L, TAING L, et al. Enrichment or depletion of a GO category within a class of genes: which test ?[J].Bioinformatics,2007,23(4):401-407.
[10] LIBERZON A, SUBRAMANIAN A, PINCHBACK R, et al. Molecular signatures database (MSigDB) 3.0[J].Bioinformatics,2011,27(12):1739-1740.
[11] BENJAMINI Y. Discovering the false discovery rate[J].Journal of the Royal Statistical Society,2010,72(4):405-416.
[12] SONG W M, ZHANG B. Multiscale embedded gene co-expression network analysis [J].Plos Computational Biology,2015,11(11),e1004574.
[13] 侯艳,杨凯,李康.基于随机森林回归的网络构建方法及应用[J].中国卫生统计,2015,32(4):558-561.
[14] LIAW A, WIENER M. Classification and regression by randomforest [J].R News,2002,2(3):18-22.
[15] SHANNON P, MARKIEL A, OZIER O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks [J].Genome Res,2003,13(11):2498-2504.
[16] 孙振球. 医学统计学[M].3版. 北京:人民卫生出版社,2014:306-313.
[17] BOLSTAD B M. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias[J].Bioinformatics,2003,19(2):185-193.
[18] CSARDI G, NEPUSZ T. The igraph software package for complex network research[J].InterJournal Complex Systems,2006,1695(5):1-9.
[19] WENG T Y, WANG C Y, HUNG Y H, et al.Differential expression pattern of THBS1 and THBS2 in Lung Cancer: clinical outcome and a systematic-analysis of Microarray Databases [J].Plos One,2016,11(8):e0161007.
[20] FISCHER H, STENLING R, RUBIO C, et al. Colorectal carcinogenesis is associated with stromal expression of COL11A1 and COL5A2 [J].Carcinogenesis,2001,22(6):875-878.
[21] ZHANG Q, BURDETTE J E, WANG J P. Integrative network analysis of TCGA data for ovarian cancer[J].BMC Systems Biology,2014,8(1):1-18.
(责任编辑: 康 锋)
Identification of Specific Genes of Cancer Stem Cells of Breast Cancer
GUOPengfei,HEPingan
(School of Sciences, Zhejiang Sci-Tech University, Hangzhou 310018, China)
Breast cancer is a kind of malignant tumor which seriously threats the health of global female. However, the hypothesis of cancer stem cell (CSC) provides a new model for breast cancer causes and treatment. In the paper, coexpression network was constructed with the bioinformatics method for 18409 genes and 1035 miRNA in 746 breast cancer samples, and they were divided into different coexpression modules. The characteristics of embryonic stem cells and mesenchymal stem cells were utilized to further screen the modules, and two gene sets related to the above two types of stem cells (size: 2019 and 859) were gained respectively. Finally, regulatory network for the two gene sets were constructed to screen specific hub genesTPX2 andMCM10 of two embryonic stem cells as well as specific geneCOL5A2 of mesenchymal stem cells. These genes can be considered as candidate specific biomarkers of CSC and potential therapeutic targets in the treatment of breast cancer.
breast cancer; embryonic stem cells; mesenchymal stem cells; gene regulatory network; hub genes
10.3969/j.issn.1673-3851.2017.05.023
2016-11-22 网络出版日期: 2017-03-28
国家自然科学基金项目(61170110, 61272312);浙江省自然科学基金项目(LY14F020049)
郭鹏飞(1990-),男,山西忻州人,硕士研究生,主要从事生物信息学方面的研究。
贺平安,E-mail:pinganhe@zstu.edu.cn
Q612
A
1673- 3851 (2017) 03- 0451- 10
猜你喜欢
今日农业(2022年13期)2022-09-15
昆明医科大学学报(2022年1期)2022-02-28
科学与社会(2021年3期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
昆明医科大学学报(2021年5期)2021-07-22
现代临床医学(2021年2期)2021-03-29
天津医科大学学报(2021年1期)2021-01-26
中国生殖健康(2020年7期)2021-01-18
中国生殖健康(2020年5期)2021-01-18
中国生殖健康(2020年3期)2020-12-19
